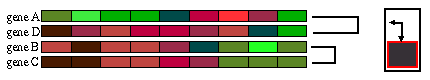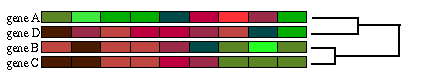|
Bioinformatics Unit - CNIO |
|
Práctica de clustering para datos de DNA arrays |
|
|
Bioinformatics Unit - CNIO |
|
Práctica de clustering para datos de DNA arrays |
|



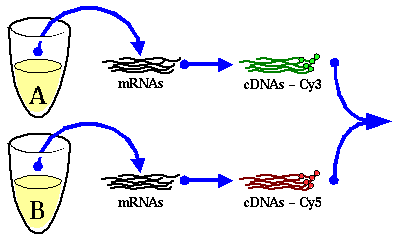
El tipo de DNA arrays más común son los cDNA arrays. Estos están formados por un cristal, un porta de vidrio, en el que se depositan unos clones en unos puntos microscópicos llamados spots. En cada cristal se pueden imprimir miles de estos spots. En cada spot encontramos muchas copias de un mismo cDNA perfectamente conocido, el cuál representará un gen en concreto.
Cada uno de los spots nos va a servir para determinar en qué medida se está expresando el gen al que representa. Para esto se extrae de la muestra que se quiere analizar el mRNA y se retrotranscribe. Durante la retrotrancripción, se marca los cDNAs que se están sintetizando con algún tipo de sonda, normalmente un fluoróforo. De este modo se podrían cuantificar todos los genes representados en el array de golpe.
Como a priori no podemos saber exactamente cuantas copias de cada cDNA hay en cada spot y para evitar problemas de saturación, se usa una referencia contra la cual se comparan los niveles de expresión de cada gen. Así, paralelamente a la extracción y tratamiento del mRNA de la muestra, se extrae y trata el mRNA de una muestra de referencia del mismo modo, salvo que se usa una sonda distinta. Típicamente, si se usan sondas fluorescentes, se elige una sonda "de otro color". Las sondas más usadas para este fin sonel Cy3 (verde) y el Cy5 (rojo).
Fig. 1.1.1 Extracción y tratamiento del mRNA: Después de extraer el mRNA de la muestra problema y de la muestra de referencia, cada una de ellas se retrotranscribe usando en el proceso al menos uno de los nucleótidos con un marcaje fluorescente distinto para cada una de las dos muestras. Luego, los dos pools de cDNA se juntan.
El siguiente paso consiste en juntar los dos pools de cDNA marcados y ponerlos en contacto con el cDNA array. En ese momento estos cDNAs se unirán a al spot que corresponda por complementariedad de bases. En cada spot se establece un proceso competitivo al final del cual quedan unidos a los cDNAs del spot más o menos cDNAs marcados de una muestra o de otra en función de la abundancia de los mismos en la mezcla de pools inicial.
A
B
Fig. 1.1.2 (A) Spot antes de la hibridación: Los cDNAs están unidos al cristal. (B) Spot después de la hibridación: los cDNAs se unen por complementariedad de bases. La cantidad de cDNA de cada una de las muestras depende de la abundancia del mismo en la mezcla de pools inicial.
Al final observaremos que cada spot aparece de un color o de otro dependiendo de si el gen que es representado por el mismo se expresa más en la muestra A o en la muestra B. Por ejemplo, si la muestra A es la de estudio y la B la de referencia, y un gen en concreto se sobreexpresa en la muestra de estudio, el spot correspondiente aparecerá más verde. Si fuera al revés, esto es, si el gen estuviera apagado o semi-apagado en la muestra de estudio, el spot aparecería más rojo. Estas diferencias se cuantifican con un escáner. Al final obtenemos para cada gen una proporción de su expresión entre una y otra muestra.
Consideremos ahora que queremos estudiar una serie de condiciones distintas (series temporales, distintas dosis de un compuesto, distintos fenotipos, etc). Para hacer esto, se repite los pasos anteriores (extracción del mRNA, retrotranscripción con incorporación de sonda fluorescente, mezcla de los dos pools de cDNA e hibridación con el cDNA array) para cada una de estas condiciones que queremos estudiar, teniendo muy en cuenta que debemos usar la misma muestra de referencia en todos los casos.
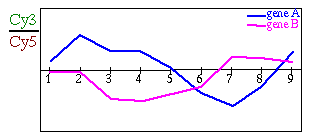
Ahora, si juntamos los datos de todos los cDNA arrays podemos averiguar en qué medida se expresa cada uno de los genes representados en el cDNA array en cada una de las condiciones estudiadas, esto es, averiguaremos el patrón de expresión de cada uno de estos genes.
A
B
Fig. 1.2.1 Distintas formas de representar los patrones de expresión (A) Patrón de expresión representado en una gráfica normal. (B) Patrón de expresión representado en una escala de colores: Es muy frecuente encontrarnos con este tipo de representación ya que resulta más clara para pintar muchos patrones a la vez.
Cuando diferentes genes presentan un mismo patrón de expresión bajo las distintas condiciones estudiadas puede ser porque desempeñan la misma función biológica, por ejemplo, para activar una ruta metabólica en concreto, se activa la transcripción de los genes implicados en esa ruta.
Así, nos va a interesar conocer cuántos tipos de patrones de expresión diferentes tenemos y qué genes están siguiendo cada uno de estos tipos de patrón.
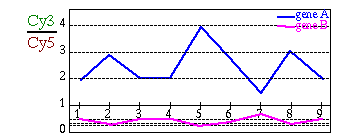
Desafortunadamente, al dividir unos valores por los otros, obtenemos valores entre 0 y 1 cuando se reprime un gen y valores entre 1 y +infinito cuando se sobreactiva. Nos gustaría tener una escala simétrica, en la que se aprecien del mismo modo las sobreexpresiones y las represiones.
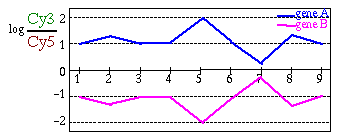
Aplicando una transformación logarítmica conseguimos este propósito. De hecho, esta es la transformación más usada. En principio cualquier otra transformación podría valer, siempre y cuando obtengamos una escala simétrica para valores de represión y de activación de los genes.
A
B
Fig. 1.4.1 Patrones de expresión: en ambos casos el gen B es el opuesto del gen A. (A) Antes de la transformación logarítmica: Todos los valores de expresión correpondientes a genes reprimidos quedan "atrapados" entre 0 y 1 y no se pueden distinguir entre sí, sólo destacan los valores de sobreexpresión. (B) Después de la transformación logarítmica: ahora pueden tener el mismo peso tanto las sobreexpresiones como las represiones.
En principio, podemos suponer que genes que comparten una misma función biológica se comportan de la misma forma. Para encontrar estas relaciones entre los genes, lo primero es saber cuándo podemos considerar que dos genes se comportan igual y cuándo no. Dicho de otro modo, debemos definir una distancia entre patrones.
Según la distancia que usemos para agrupar los distintos patrones de expresión, obtendremos unos grupos u otros, es decir, la distancia define la relación que buscamos entre los genes.
Podemos agrupar las distintas funciones de distancia en dos grandes grupos: las distancias euclídeas y las basadas en correlaciones. Las primeras se basan en diferencias absolutas mientras que las segundas se fijan en las tendencias. Si usamos una distancia euclídea tal cual, encontraremos genes en los que sus niveles de transcripción son parecidos, mientras que si miramos correlaciones, juntaremos los genes según sus tendencias.
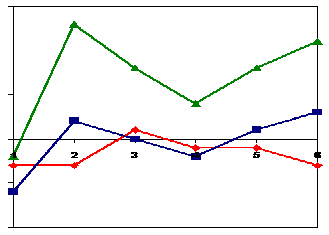
Fig. 1.5.1 Distintas distancias entre distintos patrones: si medimos distancias euclídeas, los patrones rojo y azul aparecerán juntos y el verde separado mientras que si medimos correlaciones, el verde y el azul se juntarán y el rojo no.
Según se ha visto es siempre más interesante buscar correlaciones, esto es, tendencias equivalentes, que no diferencias abslotuas. En algunos casos sin embargo se usa una distancia euclídea, pero sólo cuando previamente se han estandarizado los patrones.
Para estandarizar los patrones, basta con restar a cada valor la media del patrón y dividirlo por la desviación estándar, consiguiendo que todos los patrones estén incluidos en el mismo rango de valores. A efectos prácticos, el calcular correlaciones entre los patrones sin estandarizar o el calcular distancias absolutas entre los patrones estandarizados viene a ser lo mismo.
Se han usado diversos procedimientos para estudiar los clusters de genes cuya expresión es similar. Podemos clasificarlos en jerárquicos y no jerárquicos, según nos produzcan una clasificación basada en forma de árbol binario o nos den información acerca de posibles relaciones jerárquicas entre los datos.
El primer algoritmo utilizado fue el UPGMA, un algoritmo jerárquico aglomerativo. Los algoritmos aglomerativos son los que van juntando poco a poco los patrones más similares hasta acabar de construir el árbol completo.
A
B
C
D
Fig. 1.6.1 Clustering jerárquico clásico: se calcula la matriz de distancias entre todos los patrones (recuadro negro) y se juntan los dos elementos menos distantes, esto es, los más parecidos (recuadro rojo). (A) Los dos patrones más parecidos corresponden a los genes B y C. (B) Los genes B y C forman un cluster y se recalcula la matriz de distancias entre todos los patrones, pero esta vez en vez de considerar los genes B y C por separado, se usa la media de ambos. Ahora son los genes A y D los más próximos. (C) Los genes A y D también forman un cluster. Nótese que para conservar la jerarquía es necesario reordenar los patrones. Se vuelve a calcular la matriz de distancias considerando ya sólo los dos grupos que nos quedan. (D) Una vez que se han juntado todos los elementos se acaba el proceso.
También usaremos el SOTA (Self-Organizing Tree Algorithm), una red neuronal con topología de árbol binario. Es un método divisivo, esto es, que va separando los distintos patrones paso a paso. También se pueden usar algoritmos no jerárquicos como el SOM (Self-Organizing Maps).
| CNIO, Unidad de Bioinformática. Abril de 2002 |