Localización y enmascaramiento de secuencias repetidas. Métodos de comparación (métodos Extrínsecos o Comparative methods). Análisis de la secuencia de ADN a nivel de nucleótido (Content-based methods). Análisis de señales o motivos (Signal-based methods). Busqueda en bases de datos de secuencias expresadas.
Especialmente en el caso de genomas eucariotas, la primera fase del proceso de predicción de genes es localizar zonas de secuencias repetidas, en las que es poco probable encontrar elementos reguladores y secuencias codificantes. Las zonas de secuencias repetidas pueden ser entonces descartadas a la hora de ejecutar otros tipos de análisis, o enmascaradas, mediante la sustitución de los caracteres que representan los cuatro nucleótidos por otro símbolo arbitrario, por ejemplo "N" o "X".Además de servir para descartar regiones de menor interés desde el punto de vista funcional, el enmascaramiento de secuencias repetidas facilita la búsqueda de secuencias similares en bases de datos de secuencias con, por ejemplo, programas de la familia de aplicaciones BLAST.
Normalmente, el enmascaramiento es realizado de forma automática por muchos programas. En BLASTN, por ejemplo, el usuario puede seleccionar distintos tipos de filtro, aunque la opción por defecto enmascara las denominadas regiones de baja complejidad mediante un programa llamado DUST. Otras opciones en BLASTN permiten enmascarar secuencias repetidas humanas (LINEs y SINEs) o secuencias especificadas por el usuario, mediante el uso de minúsculas, en la secuencia usada como "query".
Uno de los programas más usados para enmascarar secuencias repetidas en secuencias de mamíferos es RepeatMasker, que filtra tanto regiones de baja complejidad, como aquellas secuencias identificadas por su similaridad a las de una base de datos de secuencias repetidas, RepBase, mantenida por el Genetic Information Research Institute (GIRI). En promedio, el 50% de cualquier secuencia genómica humana sería reconocido como secuencias repetidas, y enmascarado, por RepeatMasker.
La búsqueda de genes o de productos génicos homólogos es uno de los métodos más antiguos y más usados para identificar secuencias codificantes y determinar la estructura de genes. Este tipo de métodos se califican como extrínsecos porque se basan en usar información procedente del estudio de otros genomas. Son, por tanto, menos eficientes en Eucariotas que en Procariotas dada la menor abundancia de información de especies evolutivamente cercanas.La familia de aplicaciones BLAST incluye programas que permiten hacer diferentes tipos de búsquedas.
Si se conoce, o se puede predecir, la secuencia del producto genico, o parte de ella, otros programas permiten usar la secuencia de aminoacidos como "query":
- BLASTN permitiria identificar genes parecidos en bases de datos de secuencias de ADN, usando la secuencia genomica como "query".
- BLASTX traduce la secuencia "query" en las seis fases de lectura posibles y hace busquedas en bases de datos de proteinas; esto permitiria identificar productos genicos parecidos a los codificados por la secuencia genomica, y serviria para identificar regiones codificantes (no intrones).
- TBLASTX traduce la secuencia "query" en las seis fases abiertas de lectura y hace busquedas en bases de datos de secuencias de ADN tambien traducidas en la seis fases de lectura posibles; esto serviria para identificar secuencias que codifican para productos parecidos a los que codifica la secuencia "query".
El programa Procrustes está diseñado para intentar deducir la estructura de un gen, en cuanto a intrones y exones, basándose en la secuencia de aminoácidos de una serie de proteínas homólogas a las que supone está codificada por el fragmento de ADN que se está analizando. La secuencia de dichas proteínas homólogas tiene que ser facilitada por el usuario del programa, que podría haberlas identificado mediante búsquedas con BLASTX, por ejemplo. Si la secuencia aminoacídica facilitada es la que codifica el fragmento de ADN, Procrustes es capaz de reconstruir la estructura del gen con un 99% de exactitud.
- BLASTP busca proteinas parecidas en bases de datos de proteinas.
- TBLASTN hace busquedas en bases de datos de secuencias de ADN traducidas en la seis fases de lectura posible.
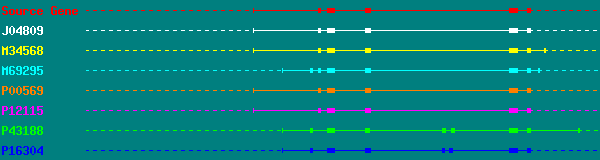
La figura de abajo es un ejemplo de resultado obtenido con Procrustes, en el que proteinas homólogas de varios organismos han sido alineadas con una secuencia genómica de ADN, para predecir la organización de intrones y exones.
El grado de conservación de cualquiera de esos motivos varía considerablemente. Aquellos relativamente conservados pueden ser identificados mediante búsquedas con secuencias consenso, que representan la secuencia del motivo para una cierta mayoría de ejemplos. Fuzznuc es un ejemplo de programa para buscar ocurrencias de una secuencia consenso en otra secuencia. El programa permite especificar el número de fallos (mismatches) que serían aceptables, así como utilizar secuencias consenso ambiguas (por ejemplo, [ACG] significa A, C o G).
Las secuencias consenso (incluso las ambiguas) tienen el problema de que no contienen información de la frecuencia con que cada nucleótido ocurre en cada posición. Dicha información puede ser expresada, sin embargo, en forma de perfiles (profiles o position weight matrices, PWMs), que son tablas en las que se recoje la frecuencia con que cada nucleótido aparece en cada posición a lo largo de la secuencia.
Una aproximación parecida es la de los algoritmos que usan
modelos
ocultos de Markov (Hidden Markov Models, HMM).
Muchos de los programas desarrollados recientemente para detectar los límites
entre exones e intrones (ver sección
3) usan esta estrategia.
Estos metodos hacen uso de informacion proviniente de tecnicas experimentales diseñadas para identificar productos genicos de forma masiva. Dos ejemplos representativos de esta estrategia son el uso de bases de datos de ESTs y de secuencias N-terminales de proteinas, que son utilizados para confirmar la existencia de genes y para deducir la estructura genica.
Los ESTs (Expressed Sequence Tags) son secuencias cortas de ADN (de 200 a 500 pb), generadas mediante la secuenciación de los extremos 5' y 3' de clones de ADNc seleccionados al azar. Los clones de ADNc se obtienen, a su vez, por transcripción reversa de ARN mensajero obtenido de una cierta fuente de células. Aunque los ADNc así obtenidos suelen corresponder con fragmentos de ARN mensajeros, la secuenciación de los extremos de los clones permite la identificación, de una manera rápida, del repertorio de genes expresados en una cierta condición, por un cierto tipo de células. Dado que las genotecas de ADNc se obtienen a partir de ARN mensajero que ha sido ya procesado, los ESTs corresponden a exones o a fragmentos de exones. Por tanto, la identificación de ESTs que corresponden con fragmentos de una secuencia de ADN genómico proporciona información sobre la localización de genes y la organización de los exones e intrones.
La principal base de datos pública sobre ESTs es dbEST, que es mantenida por el NCBI y que es, de hecho, una de las divisiones de la base de datos GenBank. Dicha base de datos almacena no sólo secuencias de ESTs, sino secuencias de ADNc completos, cuyas secuencias pueden corresponder a la práctica totalidad de los ARNm maduros que sirvieron de molde.De acuerdo al inventario de dbEST, el 15 de Marzo de 2002 había 11.018.340 entradas, de las cuales 4.200.640 correspondían a ESTs humanos y 2.547.581 a ESTs de ratón. Las entradas de la base de datos pueden ser recuperadas mediante búsquedas con palabras clave, desde la página principal de dbEST. A la hora de desarrollar el modelo de un gen, una secuencia genómica se puede usar como "query" en búsquedas con BLASTN restringidas a la base de datos de ESTs.
El EBI mantiene una página con enlaces a sitios web relacionados con ESTs (EST Links).
La identificacion de genes por busqueda en un base de datos de secuencias N-terminales de proteinas fue utilizada para anotar el genoma de Escherichia coli [Science 277:1453 (1999)].
Computational Gene Identification. R.Guigó. Curso de Doctorado en Bioinformática. UAM 2001. Computational Gene Finding. R. Guigó et al. Course 2001. Finding genes. Conceptos básicos sobre el uso de HMM para identificar genes. Protein and DNA Hidden Markov Models. User's guide for HMMer. Sean Eddy. Washington University. Predictive methods using DNA sequences. A. D. Baxevanis. En "Bioinformatics. A practical guide to the analysis of genes and proteins". Wiley-Interscience, New York 2001. Predictive methods using nucleotide sequences. J. W. Fickett. En "Bioinformatics. A practical guide to the analysis of genes and proteins". Wiley-Interscience, New York 1998. Gene prediction tools. L. Milanesi e I. B. Rogozin. En "Protein sequence analysis in the postgenomic era". CLUEB, Bologna 2001. Recent developments and future directions in computational genomics. S. Tsoka and C. A. Ouzonis. FEBS Letters 2000, 480:42. Untranslated regions of mRNAs. F. Mignone et al. Genome Biology 2002, 3(3):reviews0004.1. Páginas web de los distintos programas mencionados en, y enlazados desde, esta misma página y desde esta otra.
![]()
![]()