Verification of bond angles(Returned mail from SwissModel Modelling request)
Summary
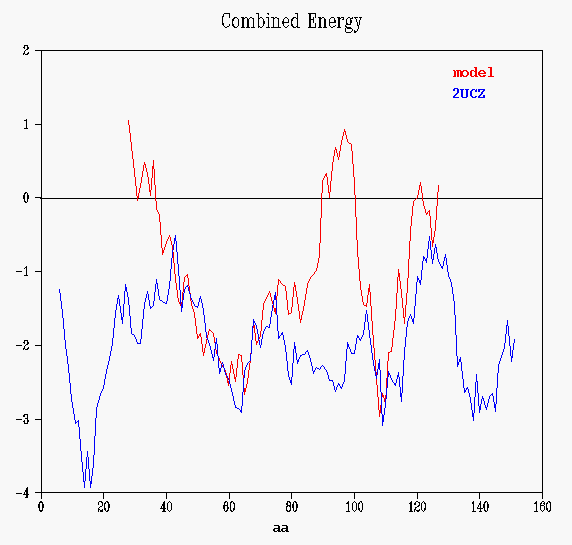
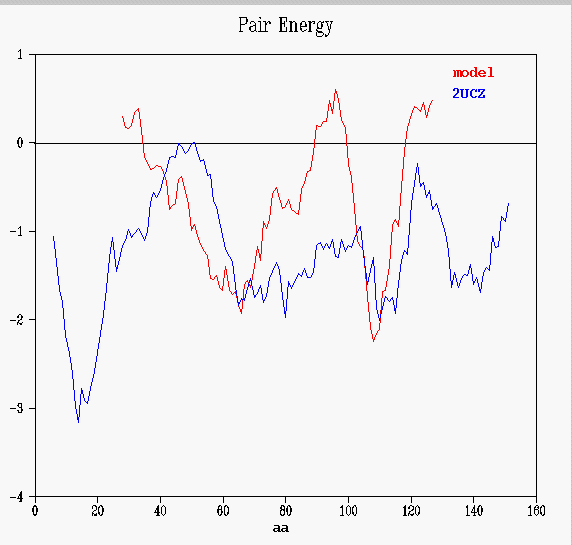
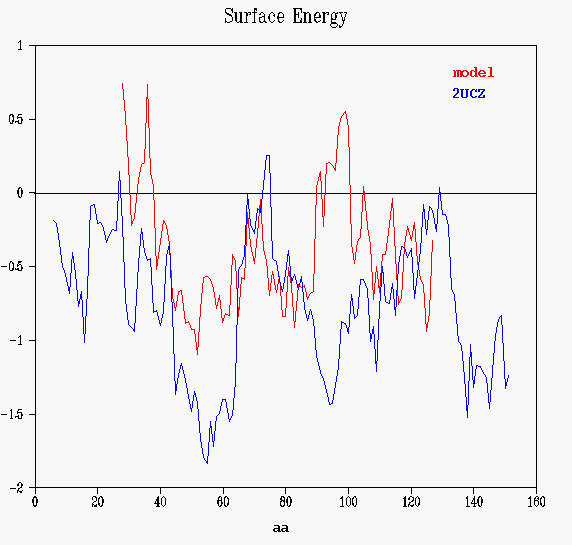
Prosa II's energy graphs provide diagnostic tools to spot problematic sections. Energy graphs display the energetic architecture of protein folds as a function of amino acid sequence position. High energies correspond to stressed or strained sections of the chain and may point to problematic parts of a fold. As long as scores and energy graphs are non native like chances are that the structure can be improved.
The total energy is a combination of pair and surface energies. The
values are smoothed by the window size set in the object. If the data are
smoothed, residue numbers refer to the central residue in the window. In
this case, winsize is 11 residues, position of the first window is 6 (that
is, the energy of each aminoacid depends on the previous-5and next-5 residues).
WhatCheck Report generated for Modelling Request AAAa006qo
Model: Batch.1.pdb
-------------------------------------------------------------
************************************************************************
********** REPORT OF PROTEIN ANALYSIS by the WHAT IF program
**********
************************************************************************
Date : 2000-02-03
This report was created by WHAT IF version 19970813-1517
INTRODUCTION
------------
This document contains a report of findings by the WHAT IF program
during the analysis of one or more proteins. It contains a separate
section
for each of the proteins that have been analysed. Each reported
fact has
an assigned severity, one of:
* error: severe errors encountered during the analyses.
Items marked
as
errors are considered severe problems requiring immediate
attention.
* warning: Either less severe problems or uncommon structural
features.
These still need special attention.
* note: Statistical values, plots, or other verbose
results of
tests
and analyses that have been performed.
If alternate conformations are present, only the first is
evaluated.
Hydrogen atoms are only included if explicitly requested, and even
then
they are not used by all checks.
Legend
------
Some notations need a little explanation:
RESIDUE: Residues in tables are normally given in 3-5 parts:
- A number. This is the internal sequence number of the
residue used
by WHAT IF.
- The residue name. Normally this is a three letter amino
acid name.
- The sequence number, between brackets. This is the residue
number
as it was given in the input file. It can be
followed by the insertion
code.
- The chain identifier. A single character. If no chain
identifier
was given in the input file, this will be invisible.
- A model number (only for NMR structures).
Z-VALUE: To indicate the normality of a score, the score may be
expressed as a Z-value or Z-score. This is just the
number of
standard deviations that the score deviates from the
expected
value. A property of Z-values is that the root-mean-square
of a
group of Z-values (the RMS Z-value) is expected to
be 1.0. Z-values
above 4.0 and below -4.0 are very uncommon. If a Z-score
is used in
WHAT IF, the accompanying text will explain how the
expected value
and standard deviation were obtained.
========================================================================
==== Compound code Model.pdb
====
========================================================================
# 1 # Error: Missing unit cell information
No SCALE matrix is given in the PDB file.
# 2 # Error: Missing symmetry information
Problem: No CRYST1 card is given in the PDB file.
# 3 # Note: No rounded coordinates detected
No significant rounding of atom coordinates has been detected.
# 4 # Note: Valine nomenclature OK
No errors were detected in valine nomenclature.
# 5 # Note: Threonine nomenclature OK
No errors were detected in threonine nomenclature.
# 6 # Note: Isoleucine nomenclature OK
No errors were detected in isoleucine nomenclature.
# 7 # Note: Leucine nomenclature OK
No errors were detected in leucine nomenclature.
# 8 # Note: Arginine nomenclature OK
No errors were detected in arginine nomenclature.
# 9 # Note: Tyrosine torsion conventions OK
No errors were detected in tyrosine torsion angle conventions.
# 10 # Note: Phenylalanine torsion conventions OK
No errors were detected in phenylalanine torsion angle conventions.
# 11 # Note: Aspartic acid torsion conventions OK
No errors were detected in aspartic acid torsion angle conventions.
# 12 # Note: Glutamic acid torsion conventions OK
No errors were detected in glutamic acid torsion angle conventions.
# 13 # Note: Heavy atom naming OK
No errors were detected in the atom names for non-hydrogen atoms.
# 14 # Warning: Chirality deviations detected
The atoms listed in the table below have an improper dihedral value
that is deviating from expected values.
Improper dihedrals are a measure of the chirality/planarity of the
structure at a specific atom. Values around -35 or +35 are expected
for chiral atoms, and values around 0 for planar atoms. Planar
side
chains are left out of the calculations, these are better handled
by the planarity checks.
Three numbers are given for each atom in the table. The first is
the Z-score for the improper dihedral. The second number is the
measured improper dihedral. The third number is the expected value
for this atom type. A final column contains an extra warning if
the
chirality for an atom is opposite to the expected value.
5 ASP ( 5 )
CA -4.1 24.6
34.0
5 ASP ( 5 )
C -7.0 -12.4
-0.1
59 VAL ( 59 )
C 6.7
12.8 -0.3
62 THR ( 62 )
C -4.3 -8.2
-0.1
64 GLY ( 64 )
C 8.4
13.6 0.1
65 GLU ( 65 )
C -4.9 -9.0
0.0
72 THR ( 72 )
C 4.6
8.6 -0.1
84 THR ( 84 )
C 5.6
10.5 -0.1
86 LEU ( 86 )
C 5.5
10.0 -0.1
98 ASN ( 98 )
C -4.4 -8.0
-0.1
# 15 # Warning: High improper dihedral angle deviations
The RMS Z-score for the improper dihedrals in the structure is
high.
For well refined structures this number is expected to be around
1.0.
The fact that it is higher than 1.5 in this structure could be
an
indication of overrefinement.
Improper dihedral RMS Z-score : 1.768
# 16 # Note: Chain names are OK
All chain names assigned to polymer molecules are unique, and all
residue numbers are strictly increasing within each chain.
# 17 # Note: Weights checked OK
All atomic occupancy factors ('weights') fall in the 0.0--1.0 range.
# 18 # Note: No missing atoms detected
All expected atoms are present.
# 19 # Note: OXT check OK
All required C-terminal oxygen atoms are present.
# 20 # Note: No extra C-terminal groups found
No C-terminal groups are present for non C-terminal residues
# 21 # Warning: Unusual bond lengths
The bond lengths listed in the table below were found to deviate
more than 4 sigma from standard bond lengths (both standard values
and sigma for amino acid residues have been taken from Engh and
Huber [REF], for DNA they were taken from Parkinson et al [REF]).
In
the table below for each unusual bond the bond length and the
number of standard deviations it differs from the normal value
is
given.
Atom names starting with "<" belong to the previous residue in
the
chain. If the second atom name is "--SS", the disulphide bridge
has
a deviating length.
5 ASP ( 5 )
N CA 1.535 4.0
7 ASP ( 7 )
C O 1.114 -5.9
8 ASP ( 8 )
N CA 1.535 4.0
14 TRP ( 14 )
CG CD2 1.346 -4.8
28 ASN ( 28 )
CG OD1 1.140 -4.5
60 ASN ( 60 )
CG ND2 1.219 -5.2
76 TRP ( 76 )
NE1 CE2 1.319 -4.6
# 22 # Note: Normal bond length variability
Bond lengths were found to deviate normally from the standard bond
lengths (values for Protein residues were taken from Engh and Huber
[REF], for DNA/RNA from Parkinson et al [REF]).
RMS Z-score for bond lengths: 0.842
RMS-deviation in bond distances: 0.017
# 23 # Warning: Directionality in bond lengths and no X-ray cell
Comparison of bond distances with Engh and Huber [REF] standard
values for protein residues and Parkinson et al [REF] standard
values
for DNA/RNA shows a significant systematic deviation.
You have most probably seen symmetry problems earlier. Please
correct these and rerun this check to see the possible implications
on the X-ray cell axes.
# 24 # Warning: Unusual bond angles
The bond angles listed in the table below were found to deviate
more than 4 sigma from standard bond angles (both standard values
and sigma for protein residues have been taken from Engh and Huber
[REF], for DNA/RNA from Parkinson et al [REF]). In the table
below
for each strange angle the bond angle and the number of standard
deviations it differs from the standard values is given. Please
note that disulphide bridges are neglected. Atoms starting with
"<"
belong to the previous residue in the sequence.
3 LEU ( 3 )
CD1 CG CD2 99.528 -5.1
5 ASP ( 5 )
<O <C N 129.774
4.2
7 ASP ( 7 )
CA CB CG 117.901 5.3
8 ASP ( 8 )
CA CB CG 129.742 17.1
14 TRP ( 14 )
CZ2 CH2 CZ3 115.010 -5.0
14 TRP ( 14 )
CZ3 CE3 CD2 124.056 4.2
17 THR ( 17 )
CA CB CG2 117.328 4.0
26 HIS ( 26 )
CA CB CG 109.342 -4.5
26 HIS ( 26 )
CG ND1 CE1 110.064 4.5
50 PHE ( 50 )
CA CB CG 109.544 -4.3
59 VAL ( 59 )
N CA CB 103.467 -4.1
59 VAL ( 59 )
CA CB CG1 97.851 -7.4
59 VAL ( 59 )
CA CB CG2 117.480 4.1
63 THR ( 63 )
CA C O 128.401 4.5
106 GLU ( 106 ) CB
CG CD 120.610 4.7
# 25 # Note: Normal bond angle variability
Bond angles were found to deviate normally from the mean standard
bond angles (normal values for protein residues were taken from
Engh and Huber [REF], for DNA/RNA from Parkinson et al [REF]).
The
RMS Z-score given below is expected to be around 1.0 for a normally
restrained data set, and this is indeed observed for very high
resolution X-ray structures. More common values are around
1.55
RMS Z-score for bond angles: 1.273
RMS-deviation in bond angles: 2.328
# 26 # Error: Side chain planarity problems
The side chains of the residues listed in the table below contain
a
planar group that was found to deviate from planarity by more than
4.0 times the expected value. For an amino acid residue that
has a
side chain with a planar group, the RMS deviation of the atoms
to a
least squares plane was determined. The number in the table is
the
number of standard deviations this RMS value deviates from the
expected value (0.0).
60 ASN ( 60 ) 9.945
8 ASP ( 8 )
7.299
106 GLU ( 106 ) 4.197
69 ASP ( 69 ) 4.147
# 27 # Note: Atoms connected to aromatic rings OK
All of the atoms that are connected to planar aromatic rings in
side
chains of amino-acid residues are in the plane within expected
RMS
deviations.
# 28 # Note: PRO puckering amplitude OK
Puckering amplitudes for all PRO residues are within normal ranges.
# 29 # Warning: Unusual PRO puckering phases
The proline residues listed in the table below have a puckering
phase
that is not expected to occur in protein structures. Puckering
parameters were calculated by the method of Cremer and Pople
[REF]. Normal PRO rings approximately show a so-called envelope
conformation with the C-gamma atom above the plane of the ring
(phi=+72 degrees), or a half-chair conformation with C-gamma below
and C-beta above the plane of the ring (phi=-90 degrees). If phi
deviates strongly from these values, this is indicative of a very
strange conformation for a PRO residue, and definitely requires
a
manual check of the data.
42 PRO ( 42 ) -55.7 half-chair
C-beta/C-alpha (-54 degrees)
61 PRO ( 61 ) 107.2 envelop
C-beta (108 degrees)
# 30 # Warning: Torsion angle evaluation shows unusual residues
The residues listed in the table below contain bad or abnormal
torsion angles.
These scores give an impression of how ``normal'' the torsion
angles in protein residues are. All torsion angles except omega
are
used for calculating a `normality' score. Average values and
standard deviations were obtained from the residues in the WHAT
IF
database. These are used to calculate Z-scores. A residue
with a
Z-score of below -2.0 is poor, and a score of less than -3.0 is
worrying. For such residues more than one torsion angle is
in a
highly unlikely position.
42 PRO ( 42 ) -2.8499
22 PRO ( 22 ) -2.5693
8 ASP ( 8 ) -2.3729
64 GLY ( 64 ) -2.3377
54 ILE ( 54 ) -2.0387
# 31 # Warning: Backbone torsion angle evaluation shows unusual
conformations
The residues listed in the table below have abnormal backbone torsion
angles.
Residues with ``forbidden'' phi-psi combinations are listed, as
well as residues with unusual omega angles (deviating by more than
3 sigma from the normal value). Please note that it is normal if
about 5 percent of the residues is listed here as having unusual
phi-psi combinations.
7 ASP ( 7 ) Poor phi/psi
8 ASP ( 8 ) Poor phi/psi
22 PRO ( 22 ) Poor phi/psi
28 ASN ( 28 ) Poor phi/psi
41 TYR ( 41 ) PRO omega poor
42 PRO ( 42 ) Poor PRO-phi
63 THR ( 63 ) Poor phi/psi, omega
poor
64 GLY ( 64 ) Poor phi/psi, omega
poor
74 ARG ( 74 ) omega poor
99 LYS ( 99 ) Poor phi/psi
101 LEU ( 101 ) Poor phi/psi, omega poor
# 32 # Note: Ramachandran Z-score OK
The score expressing how well the backbone conformations of all
residues
are corresponding to the known allowed areas in the Ramachandran
plot is
within expected ranges for well-refined structures.
Ramachandran Z-score : -1.726
# 33 # Note: Omega angle restraint OK
The omega angles for trans-peptide bonds in a structure is
expected to give a gaussian distribution with the average around
+178 degrees, and a standard deviation around 5.5. In the current
structure the standard deviation agrees with this expectation.
Standard deviation of omega values : 6.190
# 34 # Note: chi-1/chi-2 angle correlation Z-score OK
The score expressing how well the chi-1/chi-2 angles of all residues
are corresponding to the populated areas in the database is
within expected ranges for well-refined structures.
chi-1/chi-2 correlation Z-score : -0.881
# 35 # Note: Inside/Outside residue distribution normal
The distribution of residue types over the inside and the outside
of the
protein is normal.
inside/outside RMS Z-score : 1.073
# 36 # Error: Abnormally short interatomic distances
The pairs of atoms listed in the table below have an unusually
short distance.
The contact distances of all atom pairs have been checked. Two
atoms are said to `bump' if they are closer than the sum of their
Van der Waals radii minus 0.40 Angstrom. For hydrogen bonded pairs
a tolerance of 0.55 Angstrom is used. The first number in
the
table tells you how much shorter that specific contact is than
the
acceptable limit. The second distance is the distance between the
centers of the two atoms.
The last text-item on each line represents the status of the atom
pair. The text `INTRA' means that the bump is between atoms
that
are explicitly listed in the PDB file. `INTER' means it is an
inter-symmetry bump. If the final column contains the text 'HB',
the bump criterium was relaxed because there could be a hydrogen
bond. Similarly relaxed criteria are used for 1--3 and 1--4
interactions (listed as 'B2' and 'B3', respectively). If the last
column is 'BF', the sum of the B-factors of the atoms is higher
than 80, which makes the appearance of the bump somewhat less
severe because the atoms probably aren't there anyway.
Bumps between atoms for which the sum of their occupancies is lower
than one are not reported. In any case, each bump is listed in
only
one direction.
6 SER ( 6 )
N -- 14 TRP ( 14 )
CE3 0.440 2.660 INTRA
6 SER ( 6 )
N -- 14 TRP ( 14 )
CZ3 0.336 2.764 INTRA
60 ASN ( 60 )
ND2 -- 61 PRO ( 61 )
CD 0.306 2.794 INTRA BF
5 ASP ( 5 )
C -- 14 TRP ( 14 )
CE3 0.305 2.895 INTRA
67 GLN ( 67 )
CA -- 102 ARG ( 102 )
NH1 0.285 2.815 INTRA
61 PRO ( 61 )
CD -- 103 GLN ( 103 )
NE2 0.271 2.829 INTRA BF
55 ASN ( 55 )
ND2 -- 106 GLU ( 106 )
CG 0.268 2.832 INTRA
56 LEU ( 56 )
CD1 -- 93 MET ( 93 )
SD 0.237 3.163 INTRA
5 ASP ( 5 )
CA -- 14 TRP ( 14 )
CZ3 0.235 2.965 INTRA
7 ASP ( 7 )
O -- 8 ASP ( 8 )
CB 0.224 2.576 INTRA
50 PHE ( 50 )
CD1 -- 54 ILE ( 54 )
CD1 0.220 2.980 INTRA
60 ASN ( 60 )
CG -- 61 PRO ( 61 )
CD 0.209 2.991 INTRA
5 ASP ( 5 )
CA -- 14 TRP ( 14 )
CE3 0.196 3.004 INTRA
50 PHE ( 50 )
O -- 63 THR ( 63 )
CB 0.194 2.606 INTRA BF
29 ARG ( 29 )
CB -- 31 TYR ( 31 )
CE2 0.192 3.008 INTRA
59 VAL ( 59 )
CG1 -- 64 GLY ( 64 )
CA 0.186 3.014 INTRA BF
62 THR ( 62 )
C -- 64 GLY ( 64 )
N 0.159 2.741 INTRA BF
54 ILE ( 54 )
CD1 -- 59 VAL ( 59 )
CG1 0.158 3.042 INTRA
4 ALA ( 4 )
C -- 5 ASP ( 5 )
CG 0.148 3.052 INTRA
3 LEU ( 3 )
CD1 -- 17 THR ( 17 )
CB 0.140 3.060 INTRA BF
2 GLY ( 2 )
CA -- 90 ARG ( 90 )
NH2 0.136 2.964 INTRA
61 PRO ( 61 )
N -- 103 GLN ( 103 )
NE2 0.134 2.866 INTRA
50 PHE ( 50 )
CG -- 54 ILE ( 54 )
CD1 0.131 3.069 INTRA
98 ASN ( 98 )
O -- 99 LYS ( 99 )
C 0.128 2.672 INTRA
6 SER ( 6 )
O -- 8 ASP ( 8 )
N 0.122 2.428 INTRA HB
And so on for a total of 53 lines
# 37 # Warning: Abnormal packing environment for some residues
The residues listed in the table below have an unusual packing
environment.
The packing environment of the residues is compared with the
average packing environment for all residues of the same type in
good PDB files. A low packing score can indicate one of several
things: Poor packing, misthreading of the sequence through the
density, crystal contacts, contacts with a co-factor, or the
residue is part of the active site. It is not uncommon to see a
few
of these, but in any case this requires further inspection of the
residue.
23 HIS ( 23 ) -6.25
74 ARG ( 74 ) -6.11
71 HIS ( 71 ) -5.73
53 LYS ( 53 ) -5.72
102 ARG ( 102 ) -5.71
67 GLN ( 67 ) -5.30
26 HIS ( 26 ) -5.24
40 ASN ( 40 ) -5.23
41 TYR ( 41 ) -5.22
# 38 # Warning: Abnormal packing environment for sequential residues
A stretch of at least three sequential residues with a questionable
packing
environment was found. This could indicate that these residues
are part
of a strange loop, but might also be an indication of misthreading.
The table below lists the first and last residue in each stretch
found,
as well as the average residue score of the series.
100 LYS ( 100 ) --- 102 ARG ( 102 ) -4.92
# 39 # Warning: Structural average packing environment a bit worrysome
The structural average quality control value is a bit low.
The protein is probably threaded correctly, but either poorly
refined, or it is just a protein with an unusual (but correct)
structure. The average quality of 200 highly refined Xray
structures was -0.5+/-0.4 [REF].
Average for range 1 - 110 : -1.468
# 40 # Warning: Low packing Z-score for some residues
The residues listed in the table below have an unusual packing
environment according to the 2nd generation quality check. The
score
listed in the table is a packing normality Z-score: positive means
better than average, negative means worse than average. Only residues
scoring less than -2.50 are listed here. These are the "unusual"
residues in the structure, so it will be interesting to take a
special look at them.
9 ILE ( 9 ) -3.59
5 ASP ( 5 ) -3.15
23 HIS ( 23 ) -3.02
# 41 # Note: No series of residues with abnormal new packing environment
There are no stretches of four or more residues each having a quality
control Z-score worse than -1.75.
# 42 # Warning: Structural average packing Z-score a bit worrysome
The structural 2nd generation average quality control value is
a bit low.
The protein is probably threaded correctly, but either poorly
refined, or it is just a protein with an unusual (but correct)
structure. The average quality of properly refined Xray
structures is 0.0+/-1.0.
All contacts : Average = -0.517
Z-score = -3.26
BB-BB contacts : Average = 0.131
Z-score = 0.94
BB-SC contacts : Average = -0.809 Z-score
= -4.36
SC-BB contacts : Average = 0.036
Z-score = 0.39
SC-SC contacts : Average = -0.799 Z-score
= -4.09
# 43 # Note: Backbone oxygen evaluation OK
All residues for which the local backbone conformation could be
found in the WHAT IF database have a normal backbone oxygen
position.
# 44 # Warning: Unusual rotamers
The residues listed in the table below have a rotamer that is not
seen very often in the database of solved protein structures.
This
option determines for every residue the position specific chi-1
rotamer distribution. Thereafter it verified whether the
actual
residue in the molecule has the most preferred rotamer or not.
If
the actual rotamer is the preferred one, the score is 1.0. If the
actual rotamer is unique, the score is 0.0. If there are two
preferred rotamers, with a population distribution of 3:2 and your
rotamer sits in the lesser populated rotamer, the score will be
0.66. No value will be given if insufficient hits are found in
the
database.
It is not necessarily an error if a few residues have rotamer
values below 0.3, but careful inspection of all residues with these
low values could be worth it.
32 SER ( 32 ) 0.36
# 45 # Warning: Unusual backbone conformations
For the residues listed in the table below, the backbone formed
by
itself and two neighboring residues on either side is in a
conformation that is not seen very often in the database of solved
protein structures. The number given in the table is the
number of
similar backbone conformations in the database with the same amino
acid in the center.
For this check, backbone conformations are compared with database
structures using C-alpha superpositions with some restraints on
the
backbone oxygen positions.
A residue mentioned in the table can be part of a strange loop,
or
there might be something wrong with it or its directly surrounding
residues. There are a few of these in every protein, but in any
case it is worth looking at!
7 ASP ( 7 ) 0
8 ASP ( 8 ) 0
62 THR ( 62 ) 0
63 THR ( 63 ) 0
98 ASN ( 98 ) 0
99 LYS ( 99 ) 1
41 TYR ( 41 ) 2
64 GLY ( 64 ) 2
75 ASP ( 75 ) 2
# 46 # Note: Backbone conformation Z-score OK
The backbone conformation analysis gives a score that is normal
for well refined protein structures.
Backbone conformation Z-score : -1.550
# 47 # Note: Average B-factor OK
The average B-factor of buried atoms is within expected values
for
a room-temperature X-ray study.
Average B-factor for buried atoms : 12.827
# 48 # Note: Number of buried atoms with low B-factor is OK
For protein structures determined at room temperature, no more
than
about 1 percent of the B factors of buried atoms is below 5.0.
Percentage of buried atoms with B less than 5 : 0.00
# 49 # Error: The B-factors of bonded atoms show signs of over-refinement
For each of the bond types in a protein a distribution was derived
for the difference between the square roots of the B-factors of
the
two atoms. All bonds in the current protein were scored against
these distributions. The number given below is the RMS Z-score
over
the structure. For a structure with completely restrained B-factors
within residues, this value will be around 0.35, for extremely
high
resolution structures refined with free isotropic B-factors this
number is expected to be near 1.0. Any value over 1.5 is sign of
severe over-refinement of B-factors.
RMS Z-score : 3.632 over 743 bonds
Average difference in B over a bond : 4.39
RMS difference in B over a bond : 12.58
# 50 # Warning: Buried unsatisfied hydrogen bond donors and acceptors
The buried hydrogen bond donors and acceptors listed in the table
below are not involved in a hydrogen bond.
Hydrogen bond donors and acceptors that are buried inside the
protein normally form hydrogen bonds within the protein. If there
are any non hydrogen bonded buried hydrogen bond donors/acceptors
in the structure, they will be listed here.
The polar side chain atoms of ARG, LYS, GLU, ASP, HIS, ASN and GLN
are, when they are buried, almost invariably involved in at least
one hydrogen bond. If any of these atoms are listed, an
investigation should be undertaken.
9 ILE ( 9 )
N
48 VAL ( 48 )
N
52 SER ( 52 )
O
56 LEU ( 56 )
N
58 CYS ( 58 )
N
70 PHE ( 70 )
N
70 PHE ( 70 )
O
71 HIS ( 71 )
O
77 LYS ( 77 )
N
81 THR ( 81 )
N
82 MET ( 82 )
N
88 ASP ( 88 )
OD2
90 ARG ( 90 )
NE
92 GLU ( 92 )
OE2
98 ASN ( 98 )
OD1
# 51 # Note: Summary report for users of a
structure
This is an overall summary of the quality of the structure as
compared with current reliable structures. This summary is most
useful for biologists seeking a good structure to use for modelling
calculations.
The second part of the table mostly gives an impression of how well
the model conforms to common refinement constraint values. The
first part of the table shows a number of constraint-independent
quality indicators.
Structure Z-scores, positive is better than average:
1st generation packing quality : -2.421
2nd generation packing quality : -3.265 (poor)
Ramachandran plot appearance : -1.726
chi-1/chi-2 rotamer normality : -0.881
Backbone conformation
: -1.550
RMS Z-scores, should be close to 1.0:
Bond lengths
: 0.842
Bond angles
: 1.273
Omega angle restraints
: 1.125
Side chain planarity
: 2.571 (loose)
Improper dihedral distribution : 1.768 (loose)
B-factor distribution
: 3.632 (loose)
Inside/Outside distribution :
1.073
REFERENCES
==========
WHAT IF
G.Vriend,
WHAT IF: a molecular modelling and
drug design program,
J. Mol. Graph. 8, 52--56 (1990).
WHAT_CHECK (verification routines from WHAT IF)
R.W.W.Hooft, G.Vriend, C.Sander and E.E.Abola,
Errors in protein structures
Nature 381, 272 (1996).
Bond lengths and angles, protein residues
R.Engh and R.Huber,
Accurate bond and angle parameters
for X-ray protein structure
refinement,
Acta Crystallogr. A47, 392--400 (1991).
Bond lengths and angles, DNA/RNA
G.Parkinson, J.Voitechovsky, L.Clowney, A.T.Bruenger
and H.Berman,
New parameters for the refinement
of nucleic acid-containing structures
Acta Crystallogr. D52, 57--64 (1996).
DSSP
W.Kabsch and C.Sander,
Dictionary of protein secondary
structure: pattern
recognition of hydrogen bond and
geometrical features
Biopolymers 22, 2577--2637 (1983).
Hydrogen bond networks
R.W.W.Hooft, C.Sander and G.Vriend,
Positioning hydrogen atoms by optimizing
hydrogen bond networks in
protein structures
PROTEINS, 26, 363--376 (1996).
Matthews' Coefficient
B.W.Matthews
Solvent content of Protein Crystals
J. Mol. Biol. 33, 491--497 (1968).
Protein side chain planarity
R.W.W. Hooft, C. Sander and G. Vriend,
Verification of protein structures:
side-chain planarity
J. Appl. Cryst. 29, 714--716 (1996).
Puckering parameters
D.Cremer and J.A.Pople,
A general definition of ring puckering
coordinates
J. Am. Chem. Soc. 97, 1354--1358 (1975).
Quality Control
G.Vriend and C.Sander,
Quality control of protein models:
directional atomic
contact analysis,
J. Appl. Cryst. 26, 47--60 (1993).
Ramachandran plot
G.N.Ramachandran, C.Ramakrishnan and V.Sasisekharan,
Stereochemistry of Polypeptide Chain
Conformations
J. Mol. Biol. 7, 95--99 (1963).
Symmetry Checks
R.W.W.Hooft, C.Sander and G.Vriend,
Reconstruction of symmetry related
molecules from protein
data bank (PDB) files
J. Appl. Cryst. 27, 1006--1009 (1994).
![]()
![]()
Ramon Roca
Protein Design Group [Working
Page]
Centro Nacional de Biotecnología (CNB
- CSIC)
Campus Universidad Autónoma.
Cantoblanco.
28049 Madrid. Spain
Phone:+34-91-585 46 76
Fax: +34-91-585 45 06