| |




- Analysing an active site
- Building Loops
- Building a functionnal unit from a monomer
- Crystal Symmetries
- Electron Density Maps
- Energy minimisation
- Fitting Residues into Electron Density
- Homology modelling
- Making Phi/Psi statistics
- Superposing Proteins





by N.Guex &
T.Schwede
|
|
Electron Density maps : lysosyme
To complete this tutorial, you will need
to download some material (coordinates and electron density map of the lysozyme).
The data set has kindly been made available
by Dr. Doug Ohlendorf. Nate Winter did a rigid body refinement of the
coordinates deposited by K.P. Wilson, B.A. Malcom and B.W.Matthews (pdb
entry 1hel) to Doug Ohlendorf's data set.
- material for Macintosh (.hqx 986 Kb)
- material for PC (.zip 640 Kb)
The downloaded material contains 5 files:
the electron desity map (1hel.dn6), coordinates of two slightly different
models of lysozyme (1hel.pdb and 1hew.pdb), a readme, and a model of lysozyme
in which four residues have been replaced with ALA (mutant.pdb).
In this exercise, you will use the map
to identify the correct amino acid in each case replace ALA with it, and
fit it into the map.
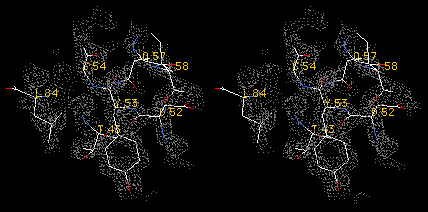
Stereo detail of a part of the lysozyme contoured at 2.0 sigma. Real-time
rendering directly from Swiss-PdbViewer.
Step by Step
- First of all, if you are using a Macintosh, make sure that Swiss-PdbViewer
has at least 6000Kb of free memory to run (click on the icon, press
Command I, and adjust the memory allocated to the program). Then launch
it.
- Open the file "1hel.pdb"
- Then, verify that the checkbox "center upon loading" of the "General
preference" is not checked. If it was checked, uncheck it, close
the protein and reload it.
- Center the view by hitting the "=" key of the numerical keypad right
mouse button on the PC), and enable the "Slab" item of the Display Menu.
- Click on the first button at the top left of the main window and set
the "slab depth" to 8Å in order to limit the quantity of information
displayed on screen at the same time.
- Select the Open DN6 Map from the file menu and load the file "1hel.dn6".
A dialog will appear. The upper part provides some information about
the unit cell size: the length along its axis, and the angle between
the axis.
- Choose the "Display around CB" radio button, set the value to 6, 6,
and 6 and contour at 1.0 sigma.
- Load the protein 1hew.pdb and make it active layer.
- Colour it by RMS. Almost everything will appear dark blue, meaning
that superposition is perfect. But one region is coloured more brightly.
- Bring up the align window, select the zone coloured by option clicking
(right mouse button on PC) and hit the return key to display only the
selected residues.
- Control click (Shift Control-Click for the PC) on the Pro70 residue
of 1hel which will center its CA in the view. Inspect the region. The
backbone is flipped. This region differs between the two pdb entries.
Colour all by B-factor, and observe that once more this is a region
with relatively high B-factor.
- Close the protein 1hew.pdb, and redisplay every amino acid of the
1hel.pdb protein.
- open the protein "mutant.pdb" As you can see, it is not superposed
onto 1hel. In fact it should be but as you have previously rotated the
protein to inspect it, the referential has changed. close the protein
"mutant.pdb", use the item "reset orientation" of the edit menu and
immediately reload the protein "mutant.pdb". Observe that the protein
is now perfectly superposed.
This illustrate the importance of the RESET orientation, that should
be used whenever you save a modification into a protein (unless the
"save in Original Orientation item of the File Menu is enabled, in which
case it will be done automatically). Otherwise when you reload it, it
will no longer fit into the electron density.
- Hide the "1hel.pdb" layer by disabling the visible box of the control
panel, and switch to the "mutant.pdb" layer.
- Center the view on Ala 38 (as you did previously with Pro70). Note
that a blob of electron density is not occupied by any atoms. Try to
figure out which residue should be put at this location (without cheating
by looking at the solved structure!). Click on the mutate tool in the
main window, click on any atom of the Ala 38, and browse the rotamer
library of your suspected residue (by hitting the * key of the numerical
keyboard). Accept the mutation of the rotamer that fits more closely
to the electron density. No rotamer fits perfectly, and you will have
to tweak the torsion angles. Click on the torsion
tool and hold down the key number 2 while clicking and moving the
mouse. It will rotate the bound binding the CB and the CD atoms. Adjust
at best the sidechain within the electron density (you might have to
use also the "1" key to rotate around the CA-CB bond. Once you are satisfied,
accept the torsion.
- Repeat the process for residues 59, 64 and 66.
Then make the "1hel.pdb" layer visible and compare your work with
the solution.
Note: in real life, after having tweaked the residues, The
atoms positions are refined again with X-PLOR,
and a new inspection of the map and tweaking is done until everything
seems to fit properly.
|
 ExPASy Home page
ExPASy Home page