I. Function
PeptIdent
is a tool that allows the identification of proteins using peptide mass
fingerprinting data.
II. Description
Peptide
mass fingerprinting involves the digestion of an unknown protein with a
proteinase of known cleavage specificity and the measurement of the resulting
peptides by mass spectrometry.
Those
experimentally measured peptide masses are then compared with the theoretical
peptides calculated for all proteins in a protein sequence database.
The
PeptIdent tool calculates the theoretical peptides of all proteins in the
Swiss-Prot/TrEMBL databases by "cutting" them with the enzyme of choice, and
calculating the theoretical masses of generated fragments. These peptides and
their masses are stored in a precomputed index. PeptIdent matches the masses of
your experimentally observed peptides with all peptide masses in this index.
Best-matching database proteins are ranked by the number of hits they have with
the observed experimental peptides.
Isoelectric
point, molecular weight and a species (or group of species) can be specified in
order to restrict the number of candidate proteins and reduce false positive
matches.
PeptIdent
makes extensive use of the annotations in Swiss-Prot/TrEMBL, and, unlike other
peptide mass fingerprinting identification programs, it takes into account
post-translational modifications as documented in Swiss-Prot. Therefore,
PeptIdent removes signal sequences and/or propeptides (as documented in the
Swiss-Prot feature table (FT lines)) before computing pI, Mw and peptide masses
for each of the resulting mature forms. The program not only returns a list of
likely protein identifications, but also any hits with peptides that are known
to carry any of more than 20 different types of discrete
post-translational modifications
.
The program thus offers a degree of protein characterization as part of the
identification procedure.
The
mass effects of several chemical protein modifications, such as oxidation of
methionine or acrylamide adducts of cysteine residues, or desired alkylation
products of cysteine residues can also be considered by the program.
 Note!
Note!
The PeptIdent tool does not consider any protein glycosylation apart from
O-GlcNAc and C-mannosylation on tryptophan. N-linked and larger O-linked sugar
structures are generally of unpredictable mass.
Note!
PeptIdent does not do any
de novo
prediction of post-translational modifications on proteins. All modified
peptides shown in the results will be the verification of an event documented
in Swiss-Prot. However, PeptIdent can match peptides whose modifications are
documented in Swiss-Prot as «potential» or «by similarity»,
and thus allows predicted post-translational modifications to be validated.
(See the document
Swiss-Prot annotation:
how is biochemical information assigned to sequence entries for the use of the terms
«potential», «by similarity» or «probable» in the
annotations.)
The
FindMod,
GlycoMod and
FindPept
tools can be used subsequently to protein identification with
PeptIdent. They allow the
- de
novo
prediction and discovery of protein post-translational modifications as well as
the prediction of potentially mutated amino acid sequences (FindMod),
- predicition of possible oligosaccharide structures occurring on proteins and
- identification of peptides that potentially result from unspecific cleavage.
PeptIdent
results are displayed on-line or can be sent to you by email, in form of an
html table. The result file contains direct links to
FindMod,
GlycoMod and
FindPept to further
characterize matching proteins by predicting potential protein post-
translational modifications and finding potential single amino acid
substitutions, to
PeptideMass and to
BioGraph for the graphical representation
of the theoretical spectrum.
III. How to use the tool
1. Name
of the unknown protein
In
the «
Name
of the unknown protein:»
text box supply a name or a code number for the query protein. For numerous
matches, give different names to each unknown protein. This is helpful if you
want to archive your query and identify it later.
2. Select
database
Under
«Database:.», select the database(s) to use for the search. You have
the choice of:
- Swiss-Prot
- Swiss-Prot
and TrEMBL
- TrEMBL
Note!
Peptides with masses >6000 Dalton are not indexed
and therefore not considered in the search.
Note!
Annotation in the TrEMBL database is done automatically; therefore it is
incomplete and not always correct. Where available, TrEMBL annotation is used
like for Swiss-Prot to process the proteins into mature chains or peptides.
TrEMBL results should therefore be interpreted with care.
Note!
Some Swiss-Prot/TrEMBL entries contain ambiguous residues (X = any amino acid,
B = Asx = Asp(D) or Asn(N), Z = Glx = Gln(Q) or Glu(E)) Examples for such
entries are
P19341,
O77721. As substitution of D by N or of Q by E induces mass
differences of about 1 Dalton, is not possible to compute exact masses for
peptides containing one or more residues B, Z or X. Those peptides are
therefore not included in the index.
3. pI and pI range
In
the «
pI:»
box, specify the pI of the protein of interest, if known. This should be
estimated from a 2-D gel. You can also specify the confidence you have in your
pI estimation by selecting the appropriate number under «
pI
range:
».
If
no number is specified, a pI of 0-
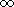
is assumed. That will cover all pI values
and return all proteins within the specified Mw region, regardless of the pI.
Note!
For bacterial proteins separated in IPG gradients, a range of ± 0.25
around the estimate is usually sufficient. For eukaryotic proteins, increase
this range to ± 0.5 units if the proteins are thought to be unmodified. If
there is a high probability that the eukaryotic protein carries
charge-modifying modifications, such as sialic acid, the range should be
changed to ± 1.
Note!
If you only have a vague idea of the protein pI, use a very large range. Even
using a pI with a large range can increase the power of your search.
Note!
pI
cannot be determined for
fragments,
i.e.
proteins
for which the sequence information in Swiss-Prot/TrEMBL is incomplete. If you
specify a pI estimate for your query, the result will include
all
fragments from the database which correspond to your other search criteria (cf.
the corresponding note for Mw of fragments.)
4. Molecular weight and Molecular weight range
In
the «
Mw»
box you should specify the estimated mass of your protein, in Dalton. This can
be estimated from a 2-D gel, or from mass spectrometry (MS) of the entire
protein. You should also specify the confidence that you have in your Mw
estimate, in percent terms, by selecting the appropriate number in the «
within
Mw range (in percent):
»
box. This allows you to limit the search to proteins within the specified
molecular weight range. If no number is specified, a molecular weight of 0-
is
assumed which means that the program searches the whole database, which
includes proteins up to a size of more than 2'000'000 Dalton (human
Titin, heart isoform,
Q10466).
Note!
For bacterial proteins larger than 20 KDa, a range of ± 20% around the Mw
estimate is usually sufficient. For small proteins, allow a +/- 40% range. For
cytoplasmic eukaryotic proteins this range is also usually sufficient, but
secreted eukaryotic proteins often carry post-translational modifications that
require a range of respectively ± 40% and 100% or more to be inclusive. If
masses have been determined with MS, the ranges used can be much smaller.
However, note that if MS has been used to determine the size of a glycoprotein
or other heavily modified protein, the measured mass this mass may be
considerably larger than the mass of the unmodified polypeptide predicted in
the database.
Note!
If you only have a vague idea of the protein Mw, you can use a large range.
However, as the proteins are ranked by the number of matching peptide masses,
very large proteins are likely to obtain a high score and appear at the top of
the list. Eliminating proteins with high molecular weight can reduce random
matches. Whenever you have an idea about the Mw range, it is highly recommended
to use this information in the identification to speed up searches and to
reduce «false positives».
Note!
Mw
cannot be determined for
fragments,
i.e.
proteins
for which the sequence information in Swiss-Prot/TrEMBL is incomplete. For
these proteins, we consider that the Mw of the complete protein is greater or
equal the molecular weight of the
known
sequence portion. Hence, if you specify a Mw estimate for your query, the
result will include all fragments from the database except those, for which the
upper boundary of your Mw range is
greater
or equal the mass of the known sequence portion from the database.
5. Species
From
the pull-down menu «
Species
to be searched:
»,
select a species or taxonomic range to limit the search to proteins from the
specified organism(s). In this case the peptide mass data is matched only
against proteins from the specified organism(s), thus eliminating many
irrelevant proteins from unrelated organisms.
To
match your peptides against peptides from all species in the database, select
"ALL". This option is not recommended without good reason, as it unnecessarily
increases the search space and causes a significant number of unrelated false
positive matches to appear.
Note!
We define "single species matching" where you, for example, have proteins from
E. coli which you then match against only the E.coli proteins in the database.
This is a good approach to use when the organism you are working with is
molecularly well defined, or ideally, the subject of a genome project.
Note!
If the source of your proteins is not molecularly well defined, it is best to
do "cross-species matching". Thus, for example, if you are working with
proteins from Candida albicans, you may wish to either match your proteins
against all proteins from fungi or against the fully sequenced yeast
Saccharomyces cerevisiae.
Note that when cross-species matching, protein pI is
frequently poorly conserved [ref], but protein mass is generally very well conserved.
You should take this into consideration when setting your pI and Mw ranges.
Note!
Peptide masses are not well conserved across species boundaries. The poor
conservation of peptide mass data is expected, as a single amino acid
substitution in any peptide can drastically change its mass [1].
Note!
Apart from a number of model organisms (e.g. human, bovine, rat, mouse, E.coli,
etc.), the pull-down menu also contains groups of species. This is useful if
working on, for example, cats, as one can match against all proteins in the
database described for mammals. If you are in doubt about the taxonomic
classification, you can consult the
NEWT Taxonomy Browser.
Note!
Proteins that are 100% conserved between different species are merged into a
single Swiss-Prot entry, e.g.
UBIQ_HUMAN,
CALM_HUMAN.
In such entries,
information about the source of each organism is noted in the OS (Organism
Species) lines, e.g. actin,
P03996:
OS Homo sapiens (Human), Mus musculus (Mouse), Rattus norvegicus (Rat),
OS Bos taurus (Bovine), and Oryctolagus cuniculus (Rabbit).
However,
the OC (Organism Classification) lines will only contain the taxonomy of the
first listed species:
OC Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Mammalia;
OC Eutheria; Primates; Catarrhini; Hominidae; Homo.
In
such cases, a PeptIdent query with RODENTIA will miss the P03996 actin entry
which describes the mouse actin sequence, but contains the organism
classification for Homo sapiens.
6. Peptide
mass data of the unknown protein
Enter
a list of peptide masses
Enter
the experimentally measured peptide masses generated from the unknown protein
in the «
Enter
a list of peptide masses...
»
text field, and separate them by
spaces, tabs or new lines.
Note!
You can copy a list of peptides from Excel or other applications and paste them
directly into the text field.
Note!
Avoid using peptide masses known to be from autodigestion of an enzyme (e.g.
trypsin!), or other artefactual peaks (e.g. matrix peaks).
Upload
a .pkm, .dta or text file
If
the peptide mass fingerprinting data is stored in a file of one of the formats
listed below, you can also upload the file directly from your computer:
(1) Click
on the on the «
Browse...»
button
(2) select
the file containing the relevant peptide mass data and
(3) click
on the «
Open»
button
The
peptide masses will then be extracted automatically from this file.
Supported
formats:
.pkm
format, produced by the Voyager software of Perseptive Biosystems or the GRAMS
software
Example:
OP=0
Center
X Peak Y Left X Right X Time X Mass Difference Name
STD.Misc
Height Left Y Right Y %Height,Width,%Area,%Quan,H/A
833.319
2189 833.260 833.378 0.016 0 0
C
0.? 0 762 762
854.843
5078 854.769 854.917 0.001 0 0
C
0.? 0 3453 3453
863.419
5108 863.064 863.775 0.001 0 0
C
0.? 0 3567 3567
872.402
12519 872.347 872.456 0.002 0 0
C
0.? 0 11417 11417
874.395
6730 874.331 874.460 0.002 0 0
C
0.? 0 3559 3559
887.786
5903 887.540 888.031 0.003 0 0
C
0.? 0 4131 4131
898.475
3329 898.416 898.534 0.006 0 0
C
0.? 0 1377 1377
904.366
7432 904.199 904.533 0.001 0 0
C
0.? 0 5596 5596
955.300
2598 955.229 955.371 0.011 0 0
C
0.? 0 1089 1089
973.845
16689 973.749 973.941 0.001 0 0
All
lines before the line containing ‘H/A’ are ignored. After that,
only lines which do not contain any capital letters in the first 20 characters
are retained. From the retained lines, the first column is interpreted as the
mass value, and the second column (if present) as the peak intensity. The
intensity is only important if the BioGraph tool is used subsequently to the
identification.
Sequest
format
Example:
1.00
1
833.319
2189
844.333
0.0
854.843
5078
863.419
5108
872.402
12519
874.395
6730
887.786
5903
898.475 3329
899.555
0.0
904.366
7432
955.300
2598
973.845
16689
The
first line is considered as a comment and is ignored. All subsequent lines are
interpreted to contain a mass and an intensity (if any), and mass values are taken into
account if the corresponding intensity is > 0.
Any
user-created files can be uploaded if they correspond to the following rules:
- The
first line does not contain any mass value (if it does, this mass value is
ignored).
- Lines
containing masses must start with the mass, and the first 20 characters must
not contain any uppercase letters.
- If
you wish to take into account peak intensities for a later visualisation of
your theoretical spectrum, the second columns of your mass lines should contain
the intensities.
Example:
>
my file
833.319
854.843
863.419
872.402
874.395
887.786
898.476
904.366
955.300
973.845
Note!
The upload option only works if you see a 'browse' button next to the text
entry field. This should be the case for most recent web browser versions, e.g.
Netscape 3.0 or higher, MS Internet Explorer 4.0 or higher.
7. Charge
state (ion mode)
You
can enter the masses of your peptides as [M] (molecular mass data),
[M-H]-
(negative ion mode, deprotonated molecular ions) or [M+H]+ (positive ion mode,
protonated molecular ions), however you must select the appropriate button.
If
you select the [M+H]+ button, all peptide masses calculated from the database
will have one proton (mass of 1 unit) added before matching with user-specified
peptides, thus giving values for singly charged peptides as found in
electrospray and MALDI-TOF mass spectrometers.
With
[M-H]- selected, all theoretical peptide masses will have one proton (one mass
unit) removed before matching.
8. Mass
type, isotopic resolution
Specify
whether the experimental mass values are «
average»
values or «
monoisotopic»
values by ticking the appropriate box. The theoretical masses of the peptides
in the database will be calculated accordingly.
9. Modification of cysteines
If
the unknown protein has been reduced and alkylated, you should specify the
reagent used for the alkylation under the "
with
cysteines treated with:
"
menu. This can be either iodoacetamide (forms carboxyamidomethyl
cysteine, Cys_CAM), iodoacetic acid (forms carboxymethyl
cysteine, Cys_CM) or 4-vinyl pyridene (forms pyridyl-ethyl
cysteine, Cys_PE).
If
the protein has not been treated in this manner, you should select the option "
nothing
(in reduced form)
"
(default setting).
Note!
Proteins are usually subject to reduction and alkylation before they are used
to generate peptides. This ensures that all disulfide bonds in a protein are
broken.
Note!
The program will modify the theoretical masses of Cys-containing peptides
accordingly, before matching with the experimental peptide masses.
10. Acrylamide
adducts on cysteines
It
is common for proteins separated by polyacrylamide gel electrophoresis that
reduced cysteines react with free acrylamide monomers. PeptIdent can therefore
be used with the option «acrylamide adducts on cysteines». With this
option selected, the program considers all cysteine residues in a peptide as
potentially modified and forming propionamide cysteine, Cys_PAM.
11. Oxidized
methionines
You
can request for all methionines in theoretical peptides to be oxidised to form
methionine sulfoxide (MSO). If this option is selected, the program will modify
the theoretical masses of all Met-containing peptides accordingly, before
matching with user-specified peptides.
Note!
Proteins separated by gel electrophoresis often show this modification.
General
note about artefactual cysteine and methionine modifications
:
If
more than one cysteine or methionine residue can be found in the peptide, the
masses of any number of possible modifications will be calculated. For example,
if there are three methionine residues in a peptide, the masses of peptides
having zero, one, two or three oxidized methionines will be calculated. The
program can also account for post-translational modifications in conjunction
with artefactual modifications in a peptide. This can be very useful, however
one should be aware that by computing all combination of possible artefactual
or post-translational modifications, a considerable amount of
«noise» is added to the database of peptide fragments. If you think
your peptides are not likely to carry artefactually modified cysteines or
methionines, it is recommended not to select any of these modifications in
order not to artificially increase the search space.
12. Mass tolerance
In
the «
Mass
tolerance: ±...
»
text box, enter the mass tolerance to be used around your peptides during
matchingSelect whether the mass tolerance is expressed as an absolute value (in
Dalton) or relative value (in parts per million; ppm).
Note!
If you have a peptide mass of 934.3 Da and specify a tolerance of ± 0.5
Da, a hit will be registered with a protein if one of its peptides in the
database has a mass between 933.8 Da and 934.8 Da.
Note!
If you have a peptide mass of 934.3 Da and specify a tolerance of ± 100
ppm, a hit will be registered with a protein if one of its peptides in the
database has a mass between 934.207 Da and 934.393 Da.
Note!
The mass tolerance should reflect the known accuracy of your mass spectrometer
(MS). Both MALDI and ES machines are now capable of achieving single decimal
point mass resolution, however, this may depend on the care that has been taken
in machine calibration and use of internal standards. We recommend the use of a
tolerance of 0.2 Da or 200ppm or better whenever it is possible.
ESI-TOF
mass spectrometers or MALDI-TOF apparatus equipped with delayed extraction and
ion reflectors are ideal for this, since most can deliver monoisotopic masses
below ±40 ppm, when two point internal calibration is used.
Less
accurate peptide mass data will require a larger mass tolerance and will result
in a lower accuracy of your search.
Note!
Mass spectrometers typically have a mass dependent error associated with mass
measurements, which cannot be uniformly expressed in Dalton. The use of ppm can
therefore be more accurate.
13. Cleavage agent
Under
«Enzyme:» you can specify the enzyme that you used to generate your
peptides. See
here for the cleavage rules.
Note!
The current version of PeptIdent only supports tryptic cleavage.
14. Missed cleavage sites
In
order to take into account partial cleavages, you can specify a maximum number
(0, 1 or 2) of missed cleavage sites to be allowed.
If
the maximum number of missed cleavages entered is 1, all concatenations of two
adjoining peptides are also added to the list of theoretical peptides under
consideration.
Note!
If you are confident that your digest was complete, with no partial fragments
present, choose the setting 0 (default setting). This will give maximum
discrimination and keep the number of random matches low.
Note!
If experience shows that your digest usually includes some peptides with missed
cleavage sites, you should specify a setting of 1, rarely 2. However, keep in
mind that each additional level of missed cleavages increases the number of
calculated peptide masses to be matched against the experimental data and the
number of random matches.
15. Minimal number of peptide matches required
Under
«
Report
only proteins with...
»
specify the minimum number of peptide mass hits you require a matching protein
to show for it to be included in the result list. The default value is 4.
Limit
the number of matching proteins displayed in the result report by selecting the
appropriate value in the «
Display
a maximum of...
»
menu. The default value is 20.
17. Print
sequence information
You
can specify if you would like the result to include, for each high-scoring
protein, information about the sequence portion covered by the matching
peptides. If this is selected, the protein sequence will be displayed, and all
matching peptides will be highlighted in color and upper case letters.
18. Send
result by e-mail
PeptIdent
results are displayed on-line in your browser window or can be sent by e-mail,
If
the results should be sent back to you by e-mail, tick the «
Send
the result by e-mail
»
box. In the «
Your
e-mail:»
text field you should enter the correct e-mail address (e.g. name@unknown.ch)
to where the results should be sent. The email option is recommended, in
particular for queries with a high number of peptide masses or for searches
against large sections of the database. This avoids timeouts («document
contains no data») which can occur for the on-line option: the browser
interrupts the connection with the program if the search is not terminated
after a certain time (usually about 3 minutes).
Note!
If you select the e-mail option, your information will be sent to the ExPASy
computer, which then undertakes the matching and returns the results to you by
e-mail. It operates in batch mode, which means several searches can be sent
successively, without having to wait for the result of the preceding query.
Note!
In batch mode, only a very limited number of requests can be treated
simultaneously, and your query will be queued for processing. Usually, results
are sent back within a few minutes. However, if the batch queue already
contains a number of requests, it will take longer (even up to a few hours in
the worst case) for your query to be returned. Please allow for a certain time
and do not unnecessarily resubmit your request. If you do not receive any
results, it is possible that you made an error when you specified your email
address. If you think there is a problem with the server, contact the server
administrator and specify the time of submission, details about your search
parameters and whether you got any error messages. Do not forget to specify
which of the ExPASy mirror sites you were using.
19. Start
PeptIdent
Once
you have filled in the form, click on the «
Start
PeptIdent
»
button to start the program.
If
you have made a mistake and would like all fields to be reset to their default
values, press the «
Reset»
button.
IV. Result
Output
1. Summary
of search parameters
The
top part of the page provides a summary of all user-specified search parameter,
as well as the date of the query, the database release number and current
number of entries, and a button to perform a new PeptIdent search.
2. List
of matching database entries
The
second part of the result page contains a summary of the best-matching protein
from the database, with a «quick jump» link to detailed peptide
information provided further down in the same page (see the following section).
This summary provides the following information for each of the high-scoring
proteins:
Score:
The
score or hit-rate for peptide mass fingerprinting is simply the number of
peptides that match the theoretical peptides from a database entry divided by
the total number of peptide masses specified for the search.
#
peptide matches:
Number of peptides that matched those from a database entry
AC:
Swiss-Prot/TrEMBL accession number (AC)
ID:
Swiss-Prot/TrEMBL
entry name (ID)
Description:
Name
of the matching protein (Swiss-Prot/TrEMBL DE line). If the matching sequence
results from cleavage of a larger precursor molecule, the name of the chain or
peptide (Swiss-Prot/TrEMBL FT CHAIN or PEPTIDE line) will be displayed.
pI:
theoretical
isoelectric
point of the matching protein.
Mw:
theoretical
molecular
weight of the matching protein in Dalton.
3. Details
of each match
user
mass:
Experimentally measured molecular mass of the peptide as provided by the user.
matching
mass:
Theoretical
molecular mass of the matching peptide as calculated from the database entry.
This mass may already include modification (post-translational or artefactual)
– in this case the type of modification is detailed in the
«modification» column.
Delta
mass:
Difference
between experimentally measured and theoretical peptide masses. This mass
difference is given in Da or ppm, depending on the mass tolerance unit
specified by the user.
#MC:
number of missed cleavage sites considered for the calculation of the
theoretical molecular mass of the peptide.
modification:
modification(s) of the peptide that were considered for the calculation of the
peptide mass.
The
following format is used to describe these modifications:
- If
only one (post-translational or artefactual) modification can be present, i.e.
the peptide contains only one Met,
or
only one Cys,
or
only one PTM documented in Swiss-Prot:
Mod_type:
position
(examples:
«Cys_CM: 32», or «CARB: 17»)
- If
at least two (post-translational or artefactual) modifications are possible in
the peptide:
Number
of occurrences of the modification x Mod_type
(example:
«2xCys_CM, 1xGLCNAC», or «1x METH», or «3xMSO»)
The
abbreviations used for the different types of modifications are listed
here.
position:
sequence
position of the matching peptide in the database entry. If the protein under
consideration is the result of post-translational processing into a mature
chain or peptide, the position information used corresponds to the numbering in
the underlying Swiss-Prot/TrEMBL entry, i.e. to the numbering relative to the
precursor sequence.
example:
The
position given for the N-terminal tryptic peptide ANSFLEELRPGNVER of the mature
chain ‘Protein C light chain (40-194)’ of PRTC_BOVIN (
P00745) is
40-54, and not 1-15.
peptide:
amino
acid
sequence
of the matching peptide
Delta
pI
:
difference between user specified/ estimated Mw and theoretical pI of the
matching protein in the database.
Delta
Mw
:
difference between user specified/ estimated Mw and theoretical Mw of the
matching protein in the database.
%
of sequence covered:
Percentage of the protein sequence covered by the matching peptides. To
calculate this percentage, the number of amino acids contained in at least one
of the matching peptides is divided by the length of the protein / mature
peptide.
Sequence:
The
sequence of the matching protein, printed in lower case letters. The regions of
the sequence that are matching the query peptides are highlighted in red and
printed in capital letters. The sequences can start from positions higher than
position 1. This reflects the removal of propeptides and signal sequences.
V. Links
The
PeptIdent output contains links to a number of related programs on ExPASy.
These links are available both from the on-line result page and from the html
file returned by email. Relevant input data and/or information about the
matching database entry are automatically transferred to those programs.
PeptIdent
A
new PeptIdent search can be launched directly from the result output. This
allows to
- Submit
a second search with slightly modified parameters, i.e. with modified molecular
weight or pI ranges, number of missed cleavages, taxonomic range etc.
- Resubmit
an archived query at a later stage, for later database releases. This is
particularly useful if the initial identification was unsuccessful or ambiguous.
Note
for users who wish to use BioGraph visualization tool subsequently to a second
search submitted via the «New Search» button:
If peptide masses were initially specified through the upload of a file which
also contained peak intensities (e.g. pkm or dta format), the peptide masses
from the original file will appear pasted into the mass window of the new
search form, where they can be modified if desired (e.g. peaks which are
believed to be «noise» or caused by a contaminant or autodigestion of
Trypsin can be removed). Peak intensities however, as read from the pkm or dta
file, will be transferred from the first to the second PeptIdent query as
hidden parameters. They have to be taken over from the original query and
cannot be modified for a second search. It is therefore not recommended to
modify / remove / add a mass before resubmitting data from an earlier query if
you intend to use the BioGraph link to visualize the theoretical
spectrum. Modification / removal / insertion of a mass would only be effective
for the mass value itself, whereas the peak intensities from the original
spectrum would remain unchanged. In case of insertion or deletion of a mass,
all intensities would be shifted and no longer be associated with the original
mass values. For the BioGraph tool to work correctly, it is more advisable to
apply modifications directly to the mass/intensity file, and to upload the file
again.
Furthermore,
direct links to a number of characterization / visualization tools are
available for each matching candidate protein:
The
FindMod tool can be used to predict post-translational modifications or single
amino acid substitutions. This is done by comparing experimental peptide masses
that did not match with the protein against those calculated from the assigned
protein sequence, seeking mass differences that may be due to
post-translational modifications. A number of rules are applied, trying to
determine whether a post-translational modification suggested by mass
difference is likely to occur in the peptide under consideration.
The
GlycoMod tool can be used to predict the possible oligosaccharide structures that occur on proteins from their
experimentally determined masses. The program can be used for free or derivatized oligosaccharides and for
glycopeptides.
The
FindPept tool can be used to identify peptides that result from unspecific cleavage of proteins from their experimental masses, taking
into account artefactual chemical modifications, post-translational modifications (PTM) and protease autolytic
cleavage.
The
PeptideMass tool can be used to simulate a theoretical digest of the matching
database protein
The
BioGraph tool allows a graphical and interactive representation of the
PeptIdent results. A theoretical spectrum is displayed, in which peaks
corresponding to matching and unmatching peptides are shown in different colors.
VI. Comments
The
output of a peptide query contains a list of proteins ranked by the number of
peptides shared with the unknown protein, where the correct identification for
an unknown protein is likely to be that with the largest number of peptide
«hits».
Confidence
in identification is achieved by looking for a significant difference in the
number of matching peptides between the top and second ranked protein, and a
good sequence coverage of the top ranked protein with the experimentally
determined peptides.
Peptide
mass data can represent a starting point for the examination of protein
modifications or processing: Peptides from a protein that do not match those
from a database may carry post-translational or artefactual modifications, or
may have undergone amino acid substitution or truncation.
Peptide
mass fingerprinting will rarely find matches for all peaks in a spectrum. This
is because some peptides, especially those that are large and/or very
hydrophobic, are either not extracted or not quantitatively extracted from a
gel or blot, and others are not ionised efficiently during mass spectrometry.
VII. References
Wilkins
M.R, and Williams K.L. (1997) Cross-species identification using amino acid
composition : a theoretical evaluation. J. Theor. Biol. 186, 7-15.
Last modified 14/Jun/2002 by ELG
 ExPASy Home page
ExPASy Home page