
| Key Points | ||
| Signals | Exons | Lengths |
| Start (ATG) | Single | Exons |
| STOPs (TGA,TAA,TAG) | First | Introns |
| Donor (GT) | Internal | Intergenic |
| Acceptor (AG) | Terminal | UTRs |
| Key Points | ||
| Signals | Exons | Lengths |
| Start (ATG) | Single | Exons |
| STOPs (TGA,TAA,TAG) | First | Introns |
| Donor (GT) | Internal | Intergenic |
| Acceptor (AG) | Terminal | UTRs |
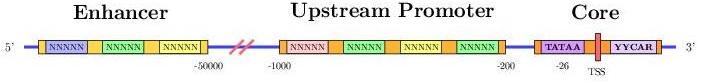
Promoter regions, upstream of genes, are made of short sequence units (termed promoter elements) which are binded by transcription factors (proteins) that modulate the transcription step. Their prediction, if accurate, would aid the prediction of genes. However, current methods are not reliable for highthroughput annotation of promoter regions.
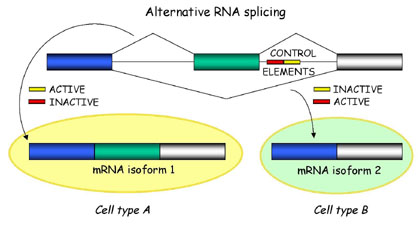
Recent data indicates that 40-50% of human genes produce alternative mRNA forms through the splicing process. How do you think current gene prediction results are effected by this fact? do you think predicted gene models can have exons which are never found together in the mRNA molecule?
Did you know that the same mRNA can have two or more translation initiation sites (ATG)? or even different polyadenilation sites? or that codon meaning can be recoded? or that frameshifting exist? However, in the light of current data, these events seem to be rare (but bear in mind that alternative splicing was considered as rare few years ago). As an example, we will study further a recoding process that happens in a peculiar family of proteins that incorporate selenium: selenoproteins.