Predicción
de las características 1D a partir de la secuencia
Actualmente no existen métodos capaces de a partir de su secuencia predecir la estructura 3D de una proteína. Sin embargo, si que se dispone de métodos capaces de predecir aspectos mas sencillos de su estructura, a partir de los cuales se puede derivar cierta información sobre su posible función. Como consecuencia de los proyectos de secuenciación de genomas iniciados en los últimos años se ha incrementado el volumen de las bases de datos de secuencias (SWISS-PROT) aunque no así el de bases de datos de estructuras (PDB).
Se ha hipotetizado, y ha sido verificado para muchas proteínas, que la estructura 3D de una proteína (esto es su plegamiento) viene determinada únicamente por la especificidad de la secuencia. Por otro lado se sabe que las proteínas chaperonas juegan a menudo un papel fundamental en el plegamiento, y aun así se asume generalmente que la estructura final es la que representa el mínimo de energía libre. Es por esto que se afirma que toda la información sobre la estructura nativa de una proteína esta codificada en su secuencia amino acídica, aunque es especifica del medio en solución en que se encuentre. Sin embargo en la practica, la falta de precisión en determinación de los parámetros básicos de los que se derivaría la estructura 3D y los recursos informáticos limitados hacen que los métodos predicción mas fiables sean aquellos basados en el conocimiento, combinación de métodos estadísticos y empíricos. Sin embargo, como se ha demostrado en los experimentos de CASP (de los que os hablaran en próximas lecturas) no se puede aun predecir estructura a partir de secuencia. Una simplificación del problema de predicción de estructura 3D es su proyección en cadenas de asignaciones estructurales. Por ejemplo, podemos asignar estados de estructura secundaria o solvatación para cada residuo identificándolos con un símbolo. De hecho, los mayores avances en bioinformática de la ultima década se han alcanzado en el campo de la de predicción de estructura secundaria. Estos avances se han alcanzado al combinar de algoritmos matemáticos complejos con la información evolutiva disponible en las bases de datos.
Las predicciones estructurales 1D , aunque locales e incluso a veces parcialmente correctas, son a menudo útiles para obtener información sobre la función de la proteína y/o los sitios activos y para llevar a cabo predicciones de mas complejas (aspectos estructurales de mayor dimensión).
Propiedades de los residuos La información mas inmediata que podemos sacar de la secuencia de una proteína son características físico-químicas de sus residuos: hidrofobicidad, polaridad, etc. Con esto podemos generar representaciones de, por ejemplo, como varía la hidrofobicidad a lo largo de la secuencia de la proteína para tener información como zonas muy hidrofóbicas, etc que puedan luego ayudarnos en la predicción de características estructurales. Hay muchas herramientas que calculan este tipo de parámetros a partir de la secuencia. Muchas han sido dotadas de interfaces WWW que facilitan en gran medida su uso.
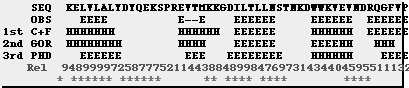
Estructura secundaria Predicción de la estructura secundaria (alpha-beta-loop) de una proteína partiendo de su secuencia de aminoácidos. La estructura secundaria se asigna generalmente de forma automática en función de su perfil de puentes de hidrogeno entre los grupos carbonilos y NH del esqueleto o "backbone". La mayoría de métodos usan redes neuronales u otros algoritmos que se entrenan con proteínas de estructura secundaria conocida para pasar luego a la predicción. Muchos de estos métodos usan información adicional proveniente, por ejemplo, de alineamientos múltiples.
Precedentes históricos de los métodos de predicción de estructura secundaria:
Una forma de introducir los métodos de predicción de estructura secundaria de proteínas es la cronológica (Eisenhaber, Persson and Argo, 1995) los describen sucintamente. Los hitos mas importantes que marcan el desarrollo de estos métodos de predicción se pueden resumir en cuatro principales:
Otra forma de introducirlos, aunque obviamente relacionada con la histórica es la basada en las características del análisis empleado, así se dividen en:
Los servidores públicos disponibles son:
El objetivo es la predicción de la exposición de un residuo al solvente. La accesibilidad se puede describir de varias formas. El método mas detallado y rápido calcula la accesibilidad estimando el volumen expuesto al solvente de cada residuo embebido en una estructura (método desarrollado por Connolly y implementado posteriormente en DSSP). Una simplificación del mismo seria pasar de los valores normalizados (el valor observado dividido por el máximo valor posible) a una descripción con dos posibles estados "buried" (accesibilidad relativa < 16%) y "exposed" (accesibilidad relativa ≥ 16%). El método clásico asignaba uno de estos dos valores "buried/exposed" en función de la hidrofobicidad del residuo, en este método zonas muy hidrofóbicas son predecidas como "buried". Sin embargo, métodos mas avanzados emplean análisis similares a aquellos empleados en la predicción de estructura secundaria (redes neuronales u otros algoritmos que se entrenan con proteínas de estructura conocida).
Entre los servidores públicos se encuentran: PHD y PROFphd (disponibles a través del servidor PredictProtein) emplean sistemas de redes neuronales que incluyen información de los alineamientos. Estos dos servidores son los únicos que predicen valores reales para la accesibilidad relativa (matriz con valores 0, 1, 4, 9, 16, 25, 36, 49, 64, 81). JPred2 emplea perfiles de PsiBlast como input para sus redes neuronales y devuelve dos estados "buried/exposed".
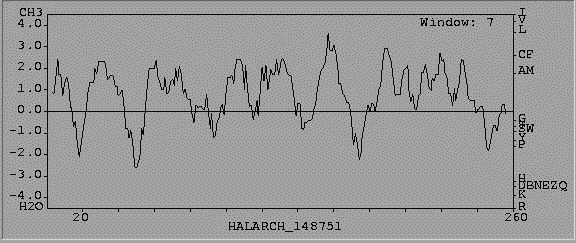
Perfil de Hidrofobicidad Kyte-Doolitle
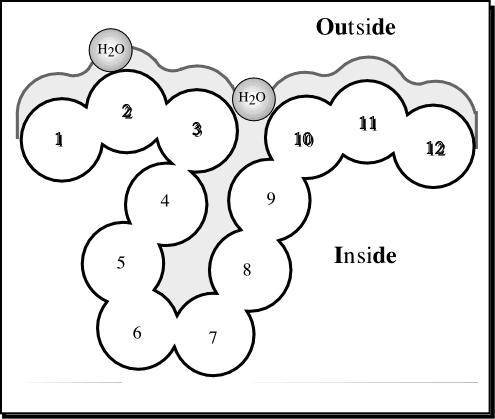
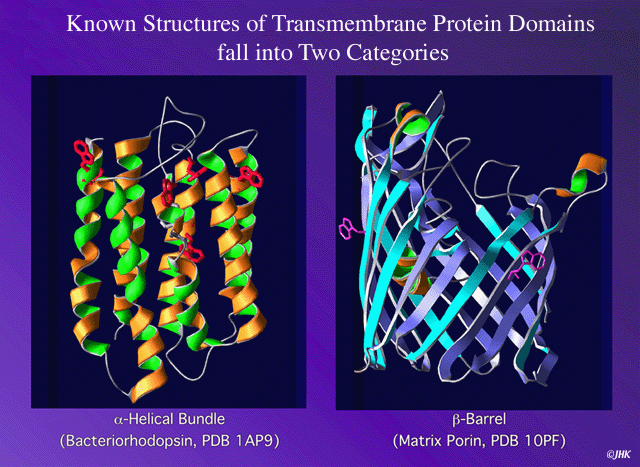
Hélices Transmembrana En el campo de la proteómica uno de los mayores retos es la determinación de la estructura de proteínas transmembrana, ya que son difíciles de cristalizar y son difícilmente analizables con NMR. Por lo tanto, la predicción de la estructura de este tipo de proteínas es de un mayor interés. Existen dos clases principales de proteínas de membrana : las que introducen hélices en la bicapa lipídica (Figura) y, proteínas que forman poros constituidos por barriles de betas (tipo porinas). Aunque recientemente han aparecido algunos servidores públicos para este segundo grupo, resulta muy dificil establecer su capacidad debido a la falta de información experimental. La situación es muy diferente para las hélice transmembrana. La estructura 3D se puede determinar conociendo la precisa localización de las hélices transmembrana explorando simplemente todas las conformaciones posibles.
A pesar de la dificultad de para su determinación experimental, estas proteínas presentan fuertes restricciones estructurales ya que la bicapa lipídica reduce los grados de libertad. Las hélices transmembrana se pueden predecir a partir de observaciones que limitan el problema: (a) estas hélices son predominantemente apolares y con una longitud de 12-35 residuos, (b) las regiones globulares entre hélices presentan típicamente longitudes menores de 60 residuos, (c) la mayoría de las hélices transmembrana tienen una distribución característica de los aminoácidos positivos arginina y lisina (definida en la regla 'positive-inside-rule' by Gunnar von Heijne) de forma que los "loops" en la zona interior de la membrana tienen mas cargas positivas que los "loops" en la zona exterior de la misma, (d) las regiones globulares largas (> 60 residuos) difieren en su composición de aquellas sujetas a la regla 'positive-inside-rule'. La mayoría de los métodos se basan en redes neuronales u otros algoritmos que se entrenan con proteínas de estructura conocida. Se consiguen mayores porcentajes de acierto ya que estas hélices suelen tener patrones muy claros (anfipaticidad, etc) que son rápidamente asimilados por los algoritmos de aprendizaje. La mayoría de los métodos compilan esta información de hidrofobicidad, y los mas avanzados incluyen la regla " positive-inside-rule" para predecir además la orientación en la membrana. La inclusión de información evolutiva mejora considerablemente las predicciones de hélices transmembrana, aunque esto se compensa de alguna forma con el crecimiento de las bases de datos de secuencias. Los servidores públicos disponibles (listado):
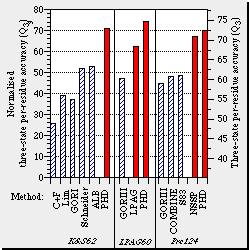
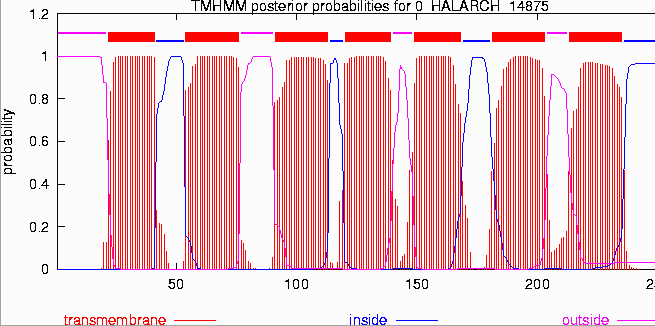
La correcta evaluación de los métodos de predicción de hélices transmembrana es difícil como consecuencia de: (a) la falta de estructuras de alta resolución que nos permitan llevar a cabo análisis estadísticos significativos, (b) los experimentos de baja resolución no proporcionan la misma calidad de información, por lo tanto no son útiles para evaluar la fiabilidad, (c) debido a la falta de datos experimentales los métodos de predicción funcionan mejor para las proteínas que se emplearon para su desarrollo. La fiabilidad de los mejores métodos (HMMTOP2, PHDhtm, y TMHMM2) predicen correctamente todas la hélices para un 70% de las proteínas estudiadas y prediciendo además en un 60% de los casos la correcta topología. La mayor fiabilidad por residuo la alcanza PHDhtm con un 70%. En general todos los métodos tienden a subestimar las predicciones en un ~86% y a confundir péptido-señal con hélices transmembrana. Además, la mayoría de los métodos, sobre todo aquellos basados solamente en escalas de hidrofobicidad, sobre-estiman las predicciones de hélices de membrana en casos de proteínas globulares en un 90%. El error de las predicciones se calcula entre un 25% para PHDhtm y un 34% para TMHMM2. Sin embargo, esto se puede contrarrestar ya que los métodos de predicción de péptidos señales son bastante fiables y la mayoría de las hélices transmembrana incorrectamente predecidas empiezan antes del décimo residuo del extremo N-terminal metionina y por lo tanto pueden ser corregidas por expertos. A pesar de los problemas de sobre-estimación, estas predicciones son útiles para buscar proteínas de membrana en genomas completos, además la mayoría no se basa exclusivamente en los valores de hidrofobocidad.
Hélices Transmembrana (Perfil TMHMM)
Modificaciones Post-transcripcionales "ExPASy Proteomics tools" (http://www.expasy.ch/tools/)
Predicción de Péptidos de Señal (SignalP) http://www.cbs.dtu.dk/services/SignalP/ Predice la presencia y localización de sitios de ruptura de péptidos señal en secuencias proteicas de diferentes organismos.
|