2 ANALISIS Y ANOTACIÓN DE GENOMAS
El análisis de genomas, una vez han sido completamente secuenciados, podría considerarse a dos niveles:Análisis estructural. Análisis funcional.
Entendemos por análisis estructural aquel que persigue hacer una descripción básica de los elementos que componen el genoma y de como están organizados.DETECCION Y MODELADO DE GENES. En el sentido mencionado mas arriba, la identificación, sobre un genoma, de la localización de genes conocidos de antemano y de genes detectados o predichos, es uno de los componentes del análisis estructural.
COMPOSICION Y PROPIEDADES FISICAS DEL GENOMA. Además, hay otra serie de parámetros medibles que se refieren más concretamente a la composición del genoma en términos de nucleótidos, como son el contenido G+C, la propensión a curvarse, la existencia de secuencias de baja complejidad.
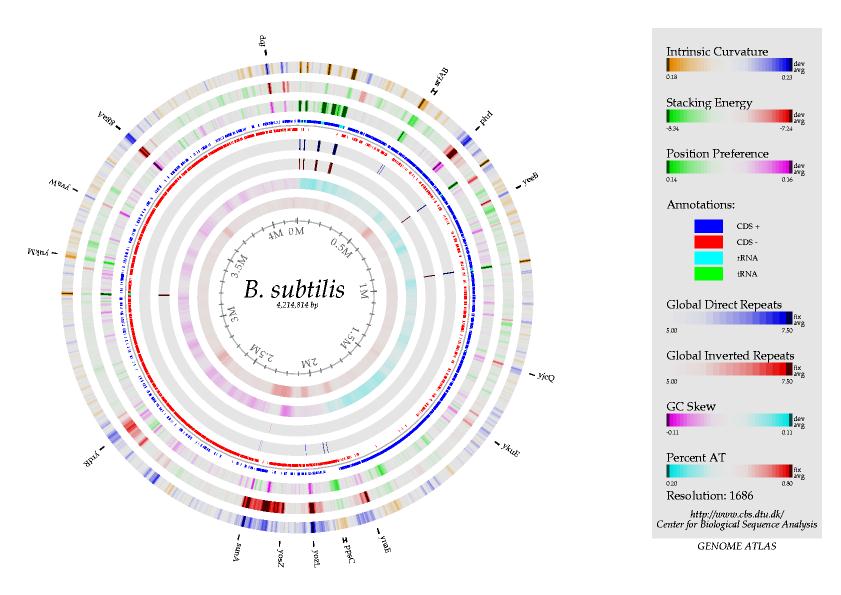
El Center for Biological Sequence Analysis (CBS) ha desarrollado un sistema automático para generar Atlas Estructurales de genomas, a partir de la información de los ficheros de secuencias del GenBank. El sistema representa, en forma de un mapa circular, una serie de parametros, entre otros, los de la siguiente lista:
En la página web de los Atlas Estructurales, el CBS mantiene una base de datos con los mapas de más de 90 genomas.Curvatura: proporcional a la propension del DNA a curvarse. Energía de stacking, cuanto más negativa, menor es la propension de las hebras del DNA a separarse. Position preference, proporcional a la rigidez del DNA. Secuencias codificantes, en las cadenas (+) y (-). Repeticiones directas Repeticiones inversas GC Skew, el contenido medio en (G-C) / (G+C) medido en una ventana deslizante de cierto tamaño. El signo de éste parametro se correlaciona con la dirección de replicación, por lo que su representación en sobre el mapa sirve para identificar los sitios de Origen y Terminación. Contenido en A+T. Los parámetros de la lista anterior son los representados en la figura de abajo, que corresponde al genoma de Bacillus subtilis.
El objetivo del análisis funcional es definir la función de cada uno de los genes de un genoma y de las proteínas que codifican, y es una de las partes más importantes del proceso de anotación de un genoma.Consideramos cuatro tipos de métodos ara predecir la función de un gen o su producto génico:
Métodos basados en detección de homología. Métodos basados en comparación de genomas. Métodos basados en el análisis de las secuencias de las proteínas. Métodos basados en la estructura de las proteínas.
Uno de los hechos más frustantes que se hicieron evidentes a mediados y finales de los años 90, cuando ya se empezaba a acumular información sobre secuencias genómicas, fué que para al menos un tercio de los genes no se conocía ni se podía predecir una función. Por otra parte, se encontró que el grado de conservación de los genes microbianos era similar entre diferentes genomas, y que alrededor de un 70% de los genes de cualquier organismo tenía homólogos en otros genomas.En base a las últimas consideraciones, fué aceptado que la función de la mayoría de los genes podría ser predicha simplemente mediante la comparación de diferentes genomas para TRANSFERIR LA ANOTACION FUNCIONAL desde proteínas de organismos mejor estudiados a sus proteinas ortólogas en organismos menos caracterizados. Actualmente, los métodos de predicción de función basados en la comparación de genomas para buscar genes homólogos comprenden las estrategias mas eficientes y más frecuentemente usadas.
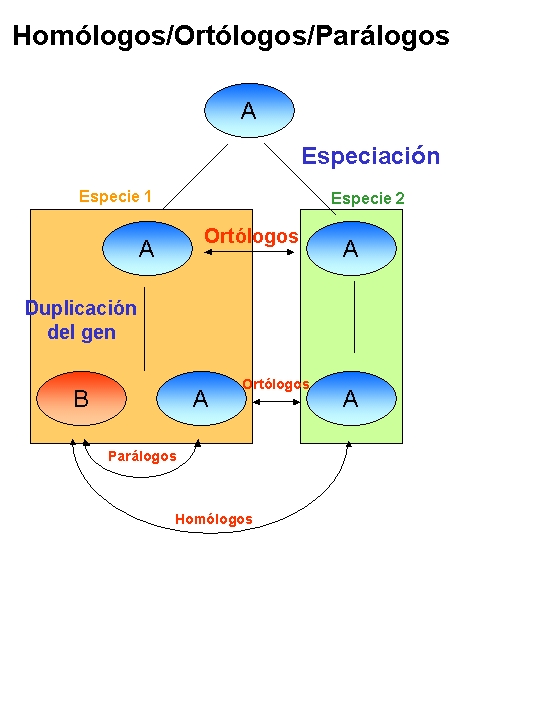
Conviene ahora recordar las diferencias entre HOMOLOGIA, ORTOLOGIA Y PARALOGIA. Dos genes, o sus correspondientes productos génicos, son HOMOLOGOS cuando poseen similitud entre si, evaluada mediante algoritmos como el de Smith-Waterman, BLAST o FASTA, (que han sido cubiertos en otra parte de este curso). Son ORTOLOGOS cuando esta similitud deriva de una ascendencia común (relación vertical), y son PARALOGOS cuando la similitud se produce dentro del mismo genoma por duplicación de un gen (relación horizontal) (figura). Se asume que dos genes ORTOLOGOS tienen la misma función, mientras que los genes PARALOGOS, puesto que se han originado por duplicación y posterior divergencia, deben de tener diferente función o, al menos, cierto grado de especialización.
A falta de una "máquina del tiempo" que permita contemplar la historia de grupos de genes homólogos, en la mayoría de los estudios se adopta una definición más práctica de ORTOLOGIA, que es la de equipararlos a los llamados Best Bidireccional Hits (BBHs).
Según esta definición, dos proteínas P1 y P2, de los organismos O1 y O2, respectivamente, son ORTOLOGAS (o BBHs) si P1 es la proteína más similar a P2 en el organismo O1, y vice versa. A esta definición se le suele imponer dos restricciones más: que la similaridad tenga una puntuación por encima de cierto valor (en el caso de BLAST suelen usarse E-values menores de 1e-5) y que las regiones de similitud entre las dos proteínas cubran, al menos, el 75% de sus longitudes.
Hay una serie de procesos evolutivos que pueden hacer que la TRANSFERENCIA DE FUNCION entre protína ORTOLOGAS no sea correcta. Entre ellos, están los siguientes:
Divergencia: a grandes distancias evolutivas, dos proteína ortólogas pueden haber divergido tanto que sea imposible reconocerlas, o diferenciarlas de proteínas parálogas. Desplazamiento no ortólogo: ocurre cuando dos genes no ortólogos realizan la misma función (por ejemplo, por un fenómeno de convergencia evolutiva). Duplicación de genes y pérdida: si dos genomas pierden diferentes parálogos de un gen ancestral que fué duplicado antes de la especiación, los genes que quedan pueden ser BBHs y no tener la misma función. Transferencia horizontal de genes: genes adquiridos recientemente pueden también impedir la identificación de los verdaderos ortólogos en el organismo receptor.
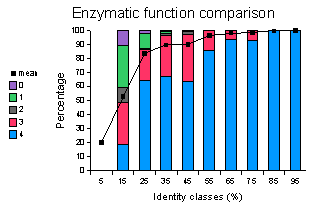
Se han hecho también estudios para evaluar la tasa de error de transferencia de función en las bases de datos existentes. En la figura siguiente se resumen los resultados de analizar, en grupos de proteínas que comparten varios grados de identidad, la frecuencia en la que tienen la misma función enzimática. Los colores corresponden al rango de ausencia de coincidencia (0) a coincidencia absoluta (4), en los cuatro digitos del codigo EC (Enzyme Classification Number; corresponden a clase, subclase, sub-subclase y enzima). De la gráfica se desprende que a partir de niveles de identidad del 35 es muy frecuente que dos enzimas tengan la misma función, o que al menos compartan la sub-subclase (Devos & Valencia, 2000).
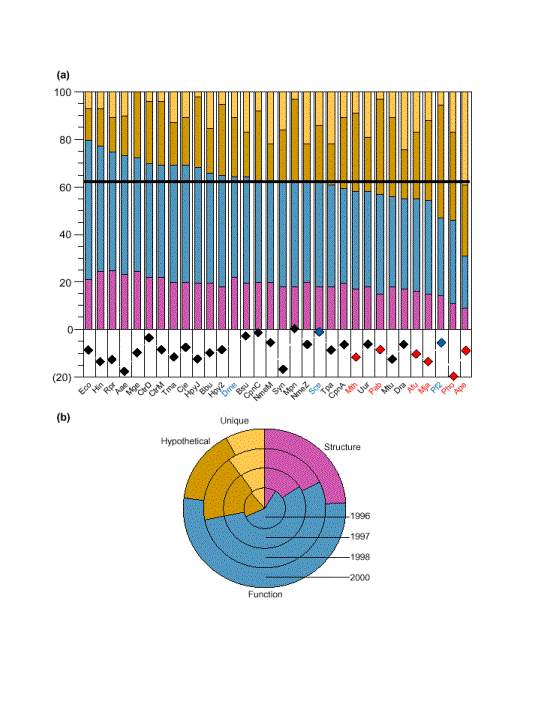
En conclusión: los sistemas de transferencia de función basados en homología parecen ser lo suficientemente sensibles. A medida que más genomas sean secuenciados, y que la actividad experimental identifique la función de nuevos genes, el número de genes únicos (sin homólogos en otros organismos) disminuirá, y el número de genes a los que se les puede asignar una función, aumentará, como se ha venido demostrando en los últimos años, aunque dependiendo del organismo, la fracción de genes de los que se desconoce la función puede llegar aún a ser mayor del 50% (figura).

Se centran en la predicción de características de los productos proteicos predichos, como por ejemplo:
Jensen et al (J Mol Biol. 2002. 319(5):1257-65) desarrollaron un sistema para predecir la función de proteínas a un nivel básico, a partir de propiedades que se pueden deducir o predecir a pàrtir de la secuencia, y lo implementaron como un servidor llamado ProtFun.- Cálculo de la composición amino-acídica y predicción de propiedades fisico-químicas.
- Presencia de motivos comunes a otras proteínas.
- Predicción de estructura secundaria.
- Presencia de regiones transmembrana y de péptidos señal.
Estos métodos se basan en la deteccion de sitios activos y en la predicción de ligandos. Aunque es una estrategia muy atractiva, de momento hay pocos ejemplos en los que se haya conseguido.
Como se ha explicado en los apartados anteriores, la principal fuente de información a la hora de predecir la función de un gen procede de búsquedas de homología, por ejemplo, con BLAST. Las predicciones basadas en el análisis de secuencias de proteinas son también usadas muy a menudo y se realizan con herramientas independientes, que utilizan un formato propio para los datos de entrada y salida. Además, muchas de estas herramientas no funcionan a traves de Internet, sino que deben ser instaladas localmente.Esta situación hace que el analizar manualmente un número grande de genes, e integrar la información obtenida, sea una tarea ingente.
Los sistemas de análisis de genomas estan diseñados para eliminar esos problemas: poseen copias locales de las herramientas y bases de datos, y agrupan el acceso a todas ellas, de modo que el usuario simplemente tiene que facilitarles la secuencia o secuencias que quiere analizar. El sistema se encarga de correr todos los programas y dar el formato adecuado a los datos para la presentación al usuario.
Como se puede suponer, los requerimentos de este tipo de sistemas son grandes: gran capacidad de almacenamiento y gran poder de cálculo (muchas secuencias pueden ser analizadas simultaneamente). Por ello, no han sido muchos los sistemas de este tipo que han sido desarrollados para el uso público.
Las páginas web de algunos sistemas de análisis de genomas se presentan mas abajo. De todos ellos, sólo GenQuiz permite a los usuarios el envío de secuencias para analizar.
![]()
{kind=link}
{kind=link}