Pairwise and Multiple Sequence Alignments,
and Similarity Searches
Links to Tools and Databases:
-
Aligning two sequences
-
BLAST
-
ClustalW
-
T-Coffee
- Databases
- Multiple Sequence Alignment viewers
Aligning
Two Sequences
>RPE_YEAST
MVKPIIAPSI LASDFANLGC ECHKVINAGA DWLHIDVMDG
HFVPNITLGQ PIVTSLRRSV
PRPGDASNTE KKPTAFFDCH MMVENPEKWV DDFAKCGADQ
FTFHYEATQD PLHLVKLIKS
KGIKAACAIK PGTSVDVLFE LAPHLDMALV MTVEPGFGGQ
KFMEDMMPKV ETLRAKFPHL
NIQVDGGLGK ETIPKAAKAG ANVIVAGTSV FTAADPHDVI
SFMKEEVSKE LRSRDLLD
>RPE_MYCPN
MLNLVVNREI AFSLLPLLHQ FDRKLLEQFF ADGLRLIHYD
VMDHFVDNTV FQGEHLDELQ
QIGFQVNVHL MVQALEQILP VYLHHQAVKR ISFHVEPFDI
PTIKHFIAQI KQAGKQVGLA
FKFTTPLVNY ERLVQQLDFV TLMSVPPGKG GQAFNSAVFN
NLKQAHKYHC SIEIDGGIKL
DNIHQIQDDV NFIVMGSGFI KLERWQRQQL LKTNQ
-
Please make a global alignment (choosing the option "needle") and also
a local alignment (choosing the option "water"). What are the differences
between the two outputs? Do you think that these two sequences are related?
-
Now try to align the two sequnces using different substitution matrices
and changing the gap opening and gap extension penalties. Use, for example,
BLOSUM62 and BLOSUM40. Do you see any difference? (you can check the pre-computed
results here).
-
How could we decide which of the alignments obtained is better?
Similarity
Search with BLAST
-
Use the sequence RPE_YEAST, to make a BLAST search on a protein database.
-
For the moment, please use the EMBL
or EBI BLAST servers, since
it is easier to retrieve the sequences identified by the BLAST search,
and we will use them later, in another exercise.
-
If you are using the EMBL BLAST server, use the following parameters:
-
database=Swiss-Prot (nrdb95 provides more
coverage, but we would obtain too many related sequences, making
the analysis more complicated).
-
filter=none
-
descriptions=250
-
alignments=250
-
Once the results of the BLAST search have been returned, you can retrieve
the sequences that have been identified as similar, by clicking on "Get
selected sequences".
-
By default, those sequences with the best p-values appear checked, but
you could select more or less sequences.
-
Those selected by default have been saved in this file.
-
Now you can try to use the NCBI
BLAST server and compare (the EMBL BLAST server uses WU-BLAST, which
is different to the original BLAST developed at the NCBI).
-
Now look for RPE_MYCPN in the output of the two BLAST searches using
RPE_YEAST as a query. Check the associated e-value (or p-value) Is the
similarity between the to sequences significant?
Note: to retrieve NCBI sequences you need to to choose the sequences you want by hand (!) and then
click on "get sequences". Then on the next page choose the two options "FASTA" and display as "text" in
the drop down menus".
Multiple
Sequence Alignment of Sequences Identified After a BLAST Search.
In this exercise you will make a multiple sequence alignment of the
sequences that have been identified in a similarity search with BLAST.
-
CREATING A MULTIPLE SEQUENCE ALIGNMENT WITH ClustalW.
- Use a ClustalW web server.
- Leave the parameters as appear by default, but change the output format:
- output format= GCG (or GCG-msf).
-
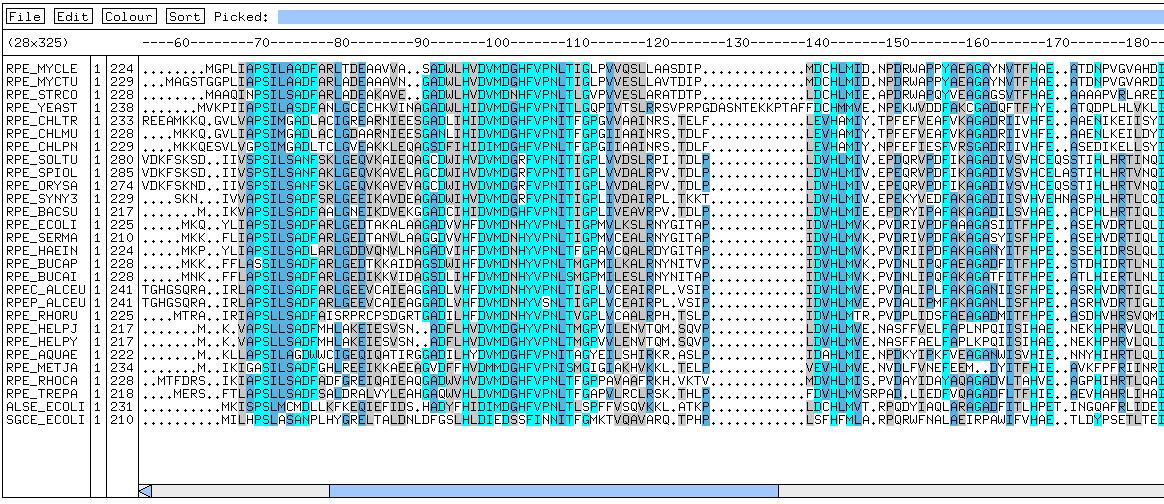
Visualisation of Multiple Sequence Alignments (MSAs)
-
Multiple sequence alignments can be more easily interpreted if the columns
in the alignment are coloured following some criteria (for example, the
degree of conservation).
-
If you are running ClustalW at the web server of the EBI,
you will have the option of getting a coloured alignment. You will have
also the options of visualizing the alignment with JalView or constructing
a tree.
{kind=link}