Familias de
proteínas.
Parte
teórica
Federico Abascal
Hasta ahora, en las
clases anteriores, en las que hemos explicado los conceptos de patrones
y perfiles, hemos utilizado frecuentemente el concepto de familia de proteínas.
De hecho hemos descrito dos bases de datos (Prosite y Pfam) que tienen
como uno de sus principales objetivos clasificar las proteínas.
También hemos
hablado de cómo evolucionan las proteínas y de que cuando
dos proteínas tienen un origen evolutivo común decimos que
son "homólogas". Hemos visto la utilidad de las herramientas del
tipo HMMer o PSI-BLAST para identificar homologías remotas.
Sin embargo, el concepto
de homólogos se queda corto cuando queremos clasificar las proteínas.
A
lo largo de la evolución, a través de procesos de duplicación
génica y divergencia, y también mediante el barajado de dominios,
aparecen nuevas subfamilias de proteínas, con nuevas funciones.
Ejemplo
hipotético de evolución de una proteína
Supongamos que en organismo
ancestral se produce una duplicación de un hipotético gen
que codifica para una proteína de secuencia:
A T F Y A G C D
E L
y supongamos que esta
proteína une e hidroliza glucosa, y que el aminoácido más
importante para reconocer la glucosa es la Cisteína de la posición
séptima.
Tras la duplicación
tendremos:
A T F Y A G C D
E L
A T F Y A G C D
E L
Tras unos "añitos",
las secuencias habrán divergido. Aquí puede ocurrir que una
de las copias degenere y acabe perdiéndose, convirtiéndose
en un pseudogén. Sin embargo, supongamos que las dos copias todavía
son capaces de dar lugar a proteínas, de secuencias:
A T F Y A G
C D E L (secuencia original)
A L
F Y A G C E E L (secuencia uno)
A S
Y Y A G C D E I (secuencia dos)
(han divergido bastante,
con respecto al original y entre sí). Supongamos que aún
son capaces de realizar la función original (o quizás una
de ellas ya no sea capaz). Supongamos ahora que en la secuencia "dos",
se produce una mutación en la séptima Cisteína, cambiando
a Glicina, y supongamos que gracias a esa mutación, la proteína
sigue siendo capaz de hacer hidrólisis pero ahora en lugar de reconocer
Glucosa, reconoce Ribosa. Ya tenemos una nueva proteína con una
nueva función. La situación es:
A T F Y A G
C D E L (secuencia original)
A L F Y A G C E
E L (secuencia uno)
A S Y Y A G G
D E I (secuencia dos)
Si es útil tener
esa nueva proteína probablemente el organismo con dicha mutación
será seleccionado positivamente. Supongamos ahora que pasan muchos
años y esta especie da lugar a otras muchas, entonces observaremos
que algunas especies habrán perdido una o las dos proteínas,
otras habrán sufrido alguna nueva duplicación, y a lo mejor
la mayoría ha conservado ambos genes, pero las secuencias habrán
continuado divergiendo.
Supongamos que tenemos
un alineamiento múltiple:
1
2 3 4 5 6 7 8 9 10
A L F Y A
G C E E L (secuencia original uno)
A L F Y A G C E
E L
A I
F R A G C E E T
A I
F R A G C E E L
A V
F Y A G C E E L
A S Y Y A
G
G D E I (secuencia
original
dos)
A S Y Y A G G
D E I
A T
Y Y
D G G D E
I
A T
Y L A G G D E
I
A S R
L A G G D E I
A S Y Y A G G
D E I
En este alineamiento
observaremos posiciones totalmente conservadas, que posiblemente tengan
que ver con la función de hidrólisis (como la sexta posición
donde hay Glicina), posiciones que son reflejo de la distinta especificidad
por ribosa o glucosa (C/G en la séptima posición), posiciones
variables y posiciones que son específicas de cada tipo de proteína
pero que no tienen que ver con la especificidad por ribosa o glucosa, sino
que reflejan la historia evolutiva (ejemplo: la posición octava
E/D).
Es decir, el hecho
de que las proteínas de tipo dos (las que unen ribosa) se
parezcan más entre sí no sólo es reflejo de las restricciones
funcionales (conservación de la séptima G), sino también
de dónde vienen (de la "secuencia original dos"; conservación
de, por ejemplo, la octava E). Las proteínas de una misma familia
o subfamilia se parecen más entre sí que con respecto a las
de otras familias o subfamilias homólogas.
La
situación actual. "Lo que vemos en las bases de datos".
La situación actual
encaja bastante bien con ese modelo hipotético: observamos que en
un grupo de proteínas homólogas en el que hay proteínas
que tienen funciones distintas, las que tienen una misma función
son más parecidas entre sí que con respecto a las otras.
Pero para describir las
relaciones entre las proteínas vemos que se nos queda corto el concepto
de homólogos, por lo que a veces hablamos en términos de
superfamilias,
familias y subfamilias y también hablamos de ortólogos
y parálogos.
Dominios
(domain shuffling)
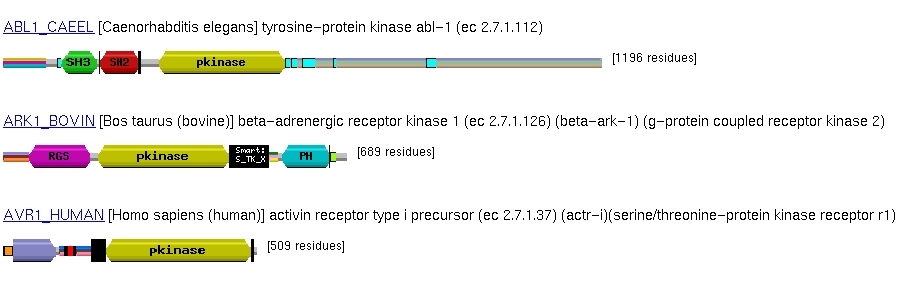
Recordemos que los dominios son unidades estructurales independientes de
las proteínas. Muchas proteínas están compuestas por
varios dominios, y sabemos que a lo largo de la evolución se ha
producido un barajado de dominios ('domain shuffling'). La función
de una proteína es el resultado de las funciones de sus dominios.
En el análisis de secuencias es importante tener en cuenta la naturaleza
multidominio de las proteínas a la hora de predecir la función
de las proteínas (hay que ser cuidadosos en la predicción
basada en homología) y a la hora de alinear proteínas homólogas.
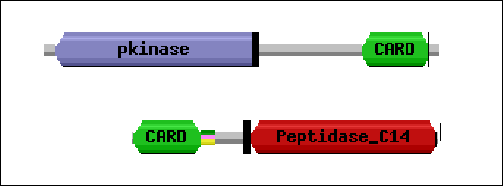
Ejemplo: proteína kinasa Rick y caspasa-9.
Los ortólogos
son aquellos genes que provienen de un mismo gen (son homólogos)
y cuya divergencia se debe a un proceso de especiación. En palabras
más sencillas, son ortólogos aquellos genes (o proteínas)
que tienen la misma identidad en distintas especies.
Por ejemplo son ortólogos
los genes de la isomerasa de glucosa-6P de Bacillus subtilis y de
Escherichia
coli.
Los parálogos
son aquellos genes cuyo último ancestro común es distinto,
es decir, la relación de ortología se ha roto (como son homólogos
sí comparten el "primer" ancestro común). Por ejemplo
las proteínas tripsina, quimiotripsina, elastasa y trombina.
Con respecto al ejemplo
que pusimos anteriormente, las proteínas de tipo dos son
ortólogas entre sí, y las del tipo uno también.
Sin embargo, las del tipo dos son parálogas respecto a las
del tipo uno.
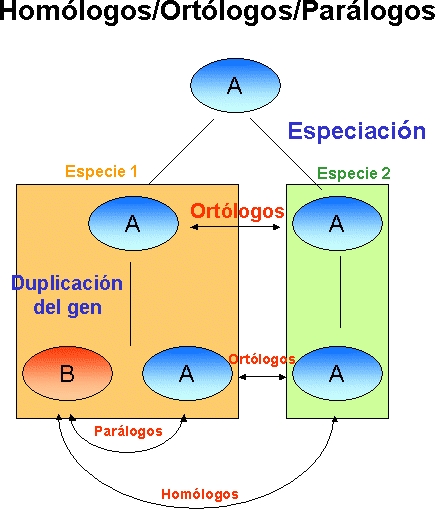
In-paralogs y out-paralogs: rara vez las relaciones evolutivas
son tan simples como en el ejemplo de los tipos uno y dos.
Muchas veces las duplicaciones no dan lugar a nuevas proteínas con
nuevas funciones, sino que los genes duplicados conservan su función
y siguen perteneciendo al grupo original. En esos casos, para referirnos
a las relaciones entre estos genes procedentes de duplicaciones recientes
se habla de in-paralogs, para distinguirlos de los otros. La razón
para hacer esta distinción es que los in-paralogs suelen conservar
las características de la proteína de la que proceden, mientras
que los out-paralogs no.
Por ejemplo, en el ser humano hay diversas copias del gen de ras,
implicado en transducción de señales. Estas proteínas
conservan más o menos la función original, aunque cada una
se expresa en distintos tejidos, bajo distintas condiciones... Son in-paralogs.
Por otra parte, las proteínas rab son parientes de
las ras, y son out-paralogs.

Superfamilias,
familias y subfamilias
Una superfamilia
es un conjunto de proteínas con un origen evolutivo común,
un conjunto de homólogos. Las superfamilias se pueden dividir, más
o menos arbitrariamente, según lo grandes que sean, en familias
y subfamilias. Son conceptos paralelos a los de ortólogos
y parálogos: las proteínas de una misma subfamilia son ortólogas
entre sí (también puede haber in-paralogs), mientras que
son parálogas de las de otras subfamilia que pertenezca a la misma
superfamilia.
|
El
interés de analizar la organización en familias de las proteínas
El objetivo más
frecuente cuando estudiamos una proteína es llegar a conocer su
función y averiguar cómo se las apaña para llevarla
a cabo. Como hemos visto, conocer cuáles son sus homólogos
nos puede ayudar, pero también es importante conocer cuáles
pertenecen a su misma familia o subfamilia, de modo que, por ejemplo, podamos
encontrar una correlación entre la conservación de determinados
residuos en la subfamilia y características funcionales específicas
de ésta. O por ejemplo, una correlación entre la organización
de dominios y las distintas funciones.
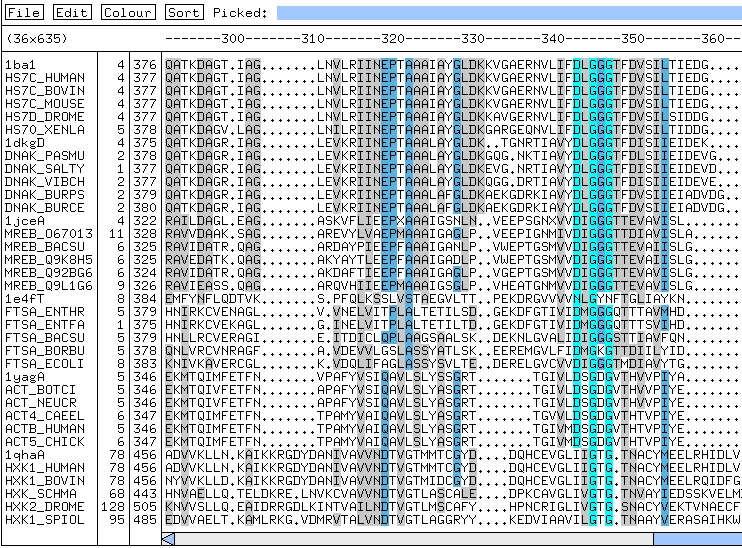
Alineamiento de proteínas de unión a ATP.
Algunos residuos están conservados en todas las familias mientras
que otros varían mucho.
Y otros presentan un patrón de conservación
dependiente de cada familia.
En la superfamilia hay: chaperones (dnak), proteínas
implicadas en la formación del septo bacteriano (ftsA, mreB), hexokinasas
(hxk), actina (act)...
La forma más frecuente
de determinas qué familias y subfamilias hay es construyendo un
árbol filogenético (esto lo veréis otro día).
El problema de los árboles es que uno tiene que buscar los homólogos,
alinearlos, construir el árbol... y, además de que esto puede
llevar bastante tiempo, a veces los árboles resultantes no son buenos,
especialmente si hay proteínas demasiado divergentes o si hay dominios
no homólogos en las proteínas que intentamos alinear. Además,
si queremos comparar dos genomas y ver qué funciones tiene uno y
cuáles el otro (quizás intentándolo correlacionarlo
con características fenotípicas de los organismos) debemos
conocer las relaciones de ortología. Y esto no podemos hacerlo manualmente
construyendo árboles para tantos genes. Por eso (entre otras razones)
existen numerosas bases de datos y métodos para estudiar la organización
de las familias de proteínas.
Las distintas bases de
datos y los distintos métodos afrontan el problema de forma diferente,
persiguiendo diversos objetivos. Unas aproximaciones tratan de encontrar
grupos de ortólogos. Otras aproximaciones, grupos de homólogos.
Etcétera.
En esta clase describiremos
algunas bases de datos de clasificación de proteínas y los
métodos que se emplean para construirlas.
Bases
de datos
Pfam
y Prosite
Estas dos bases de datos
ya las hemos explicado en la clase anterior. Se construyen a partir de
perfiles-HMM una, y a partir de patrones y perfiles simples la otra.
La limitación
básica que tienen es que no son consistentes en el nivel de definición
de los grupos: en unos casos el grupo definido en estas bases de datos
se corresponderá con una subfamilia, en otros casos con una familia
o en otros con una superfamilia. Por ejemplo, en PROSITE existe un patrón
para describir la superfamilia de las proteínas que unen ATP/GTP,
que es enorme. En Pfam, sin embargo, existen diversos dominios para las
distintas familias que unen ATP/GTP: la familia ras, la familia de factores
de elongación de la traducción, etc, etc. Por otra
parte, la familia ras de Pfam bien podría haberse subclasificado
en las subfamilias rho, rab, ran...
Sin embargo, esto frecuentemente
no resta utilidad a Pfam, que es una de las bases de datos más
empleadas. Es sólo que no afronta a fondo el problema de la clasificación
de proteínas. Una de sus ventajas es que no clasifica proteínas
sino dominios, que es la unidad evolutiva básica.
Blocks
(http://blocks.fhcrc.org/)
Podríamos dar toda una clase sobre las cosas que se pueden hacer
con BLOCKS, pero sólo daremos una visión general. Existe
un útil tutorial en BLOCKS.
Esta base de datos se construye a partir de familias descritas en InterPro
(ver más adelante) y Prints (ver más adelante). A partir
de los alineamientos de las proteínas de estas familias, se buscan
motivos
que estén conservados y que no presenten inserciones ni deleciones
(bloques; blocks). En un segundo paso, se determina cuál es el mejor
conjunto de motivos (o bloques) que definen a las proteínas de la
familia. Estos motivos se corresponden con sitios activos, sitios de unión
de substratos y cofactores y sitios con importantes implicaciones estructurales.
Esta clasificación tiene algunas ventajas porque permite que
fácilmente veamos cuáles son las pequeñas zonas conservadas
que son características de una familia. También es interesante
para determinar si una proteína de la familia carece de alguno de
los motivos.
Lo mejor de BLOCKS es el interfaz web que ofrece: permite realizar múltiples
consultas, ver los perfiles de los motivos, construir árboles, buscar
con los motivos en otras bases de datos...
Tour: buscaremos por "keyword",
introduciendo "cytosine and methylase", gracias a lo cual obtendremos la
entrada IPB001525
(se corresponde con la entrada IPR001525 de InterPro). Si hacemos click
en ella veremos: una serie de motivos IPB001525A, IPB001525B, IPB001525C,
IPB001525D, IPB001525E y IPB001525F. Podemos ver la longitud de cada uno
y algunas otras características.
Por otra parte, podemos ver la distribución de los motivos
en todas las proteínas (pinchando en Block Map).
IPB001525: C-5 cytosine-specific DNA methylase
6 distinct blocks in 158 sequences
MTA1_ARTLU|P31974 ( 521) -A-----BB-CCC---DDD--------------------------EE-F-----
MTB6_BACSP|P43420 ( 315) A-----BB-CCC--DDD----------EE-F--
MTB1_BACBR|P34905 ( 374) -A------BB-CCC--DDD-----------EE-F----
MTD2_HERAU|P25265 ( 354) -A-----BB-CCC--DDD------------EE--F---
MTA1_RUEGE|P94147 ( 429) A-----BB-CCC--DDD---------------------EE-F--
(...)
También podemos ver gráficamente los perfiles para cada motivo,
pinchando en "Logos".
O también podemos ver un árbol de las secuencias (pinchando
en ProWeb TreeViewer).
También, pinchando en "Structures" (cuando se conoce
la estructura tridimensional de alguna de las proteínas) podemos
ver la distribución de los motivos de BLOCKS en la estructura (los
motivos están coloreados; también observamos la cadena de
ADN a la que se unen estas proteínas).
Además, podemos utilizar una entrada de BLOCKS para buscar en
bases de datos de secuencias, usando alguno de estos caminos:
-
COBBLER sequence: construye una especie de secuencia consenso con
las regiones de los motivos. Supuestamente, con esta secuencia, mediante
BLAST, seremos capaces de encontrar más homólogos.
-
MAST Search: genera PSSM (perfiles) para cada motivo y permite buscar
con ellos. Aquí tenéis un resultado de ejemplo.
[MEME (Multiple Em for Motif Elicitation) & MAST (Motif alignment
and search tool) website]
-
LAMA search: es una herramienta para comparar alineamientos múltiples.
En BLOCKS se usa para comparar una entrada de BLOCKS con las otras entradas
(o con las de la base de datos Prints), de modo que podemos encontrar relaciones
de homología entre las distintas familias. Aquí tenéis
un resultado de ejemplo:
LAMA version 28 Apr 00.
Minimal length of reported alignments 4
Score cutoff is 5.6 Z score units (in the top 7.7e-05 percentile
of chance scores)
alignment Z-score expected number for
block 1 from:to
block 2 from:to length
searching 5000 blocks
IPB001525A 1 : 14 and IPB001525A
1 : 14 (14) score 100 (19.2 0.0e+00)
IPB001525A 2 : 14 and IPB001566A
3 : 15 (13) score 47 ( 6.2 1.3e-01)
IPB001525A 1 : 13 and IPB001614A
28 : 40 (13) score 45 ( 5.7 3.5e-01)
IPB001525B 1 : 16 and IPB001525B
1 : 16 (16) score 100 (21.1 0.0e+00)
IPB001525B 2 : 16 and IPB001532D
11 : 25 (15) score 41 ( 5.6 3.7e-01)
IPB001525C 9 : 27 and IPB000146D
3 : 21 (19) score 37 ( 5.7 3.5e-01)
IPB001525C 2 : 23 and IPB000352A
26 : 47 (22) score 36 ( 5.6 3.6e-01)
IPB001525C 1 : 28 and IPB001525C
1 : 28 (28) score 100 (31.8 0.0e+00)
IPB001525C 8 : 28 and IPB002857C
1 : 21 (21) score 43 ( 7.2 1.3e-02)
IPB001525D 1 : 27 and IPB001525D
1 : 27 (27) score 100 (31.2 0.0e+00)
IPB001525D 1 : 27 and IPB004000C
15 : 41 (27) score 29 ( 5.9 2.0e-01)
IPB001525E 1 : 16 and IPB001525E
1 : 16 (16) score 100 (21.1 0.0e+00)
IPB001525E 3 : 16 and IPB002584A
6 : 19 (14) score 46 ( 6.0 1.7e-01)
IPB001525E 1 : 16 and IPB002857F
16 : 31 (16) score 65 (12.1 0.0e+00)
IPB001525F 1 : 10 and IPB001525F
1 : 10 (10) score 100 (14.5 0.0e+00)
IPB001525F 1 : 10 and IPB002190D
20 : 29 (10) score 57 ( 6.5 5.5e-02)
El IPB002857
se corresponde con los "CXXC zinc finger". ¿Será esta familia
homóloga de la de IPB001525
(C-5 cytosine-specific DNA methylase), que también se une a ADN?
Sugerencias: buscar contra Pfam y ver si coinciden los hits de unos y otros;
echar un vistazo a los "logos"; hacer BLAST; ver las anotaciones funcionales;
si ambos tuviesen estructuras tridimensionales conocidas podríamos
ver si éstas se parecen; también se puede ver si las proteínas
que están presentes en un BLOCK también lo están en
el otro.
En realidad lo que ocurre en este caso
no es que haya dos familias relacionadas, sino que para definir a las metilasas
éstas se ha utilizado una serie de motivos que abarcan varios dominios.
La estructura de una de las metilasas es:

Sin embargo hay muchas otras proteínas
que no son metilasas que presentan el dominio RING-Zn finger y para todas
esas proteínas existe otra entrada en BLOCKs. Ambas entradas solapan.
Además de todo esto que podemos hacer a partir de un BLOCK, podemos
buscar con secuencias de aminoácidos o de nucleótidos.
También podemos construir BLOCKS con "Block Maker". En el tutorial
se explican estos aspectos.
Prints
(http://bioinf.man.ac.uk/dbbrowser/PRINTS/)
Aquí
tenéis un tutorial sobre PRINTS.
El enfoque de esta base de datos es similar al de BLOCKS. Agrupa las
proteínas en superfamilias, familias y subfamilias de acuerdo a
la presencia de conjuntos de pequeños motivos, a la "huella digital"
(fingerprint) que caracteriza a una familia de proteínas.
¿Cómo se construye la base de datos?: a partir de alineamientos
múltiples se derivan automáticamente perfiles para motivos
conservados. Con éstos se realizan búsquedas en las bases
de datos de secuencias. Con las nuevas proteínas se mejoran los
motivos, se construyen de nuevo perfiles para cada motivo y se realizan
nuevas búsquedas. Finalmente, se obtiene para la familia en cuestión,
una lista de motivos que caracterizan de forma óptima a la familia
y una lista de las proteínas que los presentan, indicando qué
proteínas presentan todos los motivos y cuáles sólo
algunos.
Un par de ejemplos de fingerprints: FASRECEPTOR
y IL1BCENZYME.
También existe un enlace para ver la estructura tridimensional
y dónde se localizan los motivos del 'fingerprint'.
Se pueden realizar búsquedas con FPScan
para determinar a qué fingerprints se parece una determinada secuencia.
Aquí
tenéis un ejemplo, el resultado de buscar con swiss:RASH_HUMAN.
Smart
(http://smart.embl-heidelberg.de/;
mirror en http://smart.ox.ac.uk/)
[ Smart Modular Architecture Research
Tool
]
La versión 4.9, de febrero de 2004, contiene 665 dominios.
Los dominios se construyen de forma parecida a Pfam: se elaboran HMMs semilla
para determinados dominios y con esos HMMs se busca en las bases de datos
para anotar las proteínas.
Para cada dominio se muestra información de en qué especies
está presente, con qué otros dominios aparece, la localización,
enlaces a la estructura tridimensional, etcétera. Muy parecido a
Pfam.
Quizás la característica más particular de SMART
es que pone el énfasis en definir los dominios móviles de
eucariotas, es decir, aquellos dominios más promiscuos como SH2,
SH3, dominio plecstrina, etcétera los cuales aparecen en muy diversas
combinaciones. Además ofrece un servicio de búsqueda de proteínas
según la organización de dominios que tengan. Por ejemplo,
podemos buscar todas aquellas proteínas que tengan los dominios
CARD y CASc pero que no tengan el dominio BIR.
La definición de los dominios puede variar de una base de datos
a otra. Por ejemplo, según Pfam hay 226
proteínas que tienen el dominio caspasa, mientras que en SMART son
sólo 186.
PRODOM
(y ProDom-CG)
[http://prodes.toulouse.inra.fr/prodom.html]
Hay un tutorial en http://prodes.toulouse.inra.fr/prodom/2002.1/documentation/help.php.
También es una base de datos de dominios. Su peculiaridad más
sobresaliente es que la clasificación se hace automáticamente,
usando el programa MKDOM2. La idea es que la secuencia más corta
(siempre y cuando no sea un fragmento de una proteína) se corresponderá
con un dominio. Con esa secuencia se inicia un PSI-BLAST iterativo y con
los segmentos homólogos de otras proteínas ya tenemos definido
un dominio. Estos segmentos son "eliminados" de la base de datos. Los segmentos
de esas proteínas que no sean homólogos permanecen en la
base de datos. Con la siguiente secuencia más corta se inicia otra
vez el proceso. Y así sucesivamente hasta que no queden secuencias
en la base de datos.
En las últimas versiones también se generan los dominios
a partir de los que ya están descritos en Pfam-A, y también
a partir de la base de datos SCOP.
Y también algunos dominios son corregidos manualmente por expertos.
A partir de estos dominios se realizan búsquedas PSI-BLAST del mismo
modo que con los dominios descritos automáticamente.
El resultado son 391.935 dominios (de un total de una base de datos
de unas 556.964 secuencias). Y existen 144.444 familias de proteínas
con al menos dos dominios.
Esta base de datos puede resultar útil en aquellos casos en que
no exista una definición de un dominio dado en otras bases de datos
como Pfam, Prosite o Smart.
ProDom-CG (CG: complete genome) es un subconjunto de ProDom para los
proteomas correspondientes a genomas ya secuenciados.
ProDom es la fuente de información que se utiliza para construir
el suplemento Pfam-B.
InterPro
[http://www.ebi.ac.uk/interpro/]
Hay documentación en: http://www.ebi.ac.uk/interpro/user_manual.html?
Hay un tutorial en: http://www.ebi.ac.uk/interpro/tutorial.html
Hemos visto que son muchas las bases de datos de dominios y motivos,
que cada una describe distintos niveles de la organización de las
proteínas (y muchas veces de una forma que no es consistente), que
cada una usa distintos métodos... por eso se decidió crear
InterPro, que es una base de datos que se construye a partir de Pfam,
SMART,
Prosite,
Prints,
ProDom
y TIGRFams.
Hace un año aproximadamente...
| Database |
Version |
Entries |
| SWISS-PROT |
41.0 |
122.564 |
| PRINTS |
35.0 |
1.750 |
| TrEMBL |
23.0 |
830.525 |
| Pfam |
8.0 |
5.193 |
| PROSITE patterns |
17.37 |
1.605 |
| PROSITE preprofiles |
N/A |
150 |
| ProDom |
2002.1 |
1.021 |
| InterPro |
6.0 |
7.751 |
| Smart |
3.4 |
654 |
| TIGRFAMs |
2.1 |
1.614 |
|
Actualmente...
| Database |
Version |
Entries |
| SWISS-PROT |
42.5 |
138.992 |
| PRINTS |
37.0 |
1.850 |
| TrEMBL |
25.5 |
1.013.263 |
| Pfam |
11.0 |
7.255 |
| PROSITE patterns |
18.10 |
1.659 |
| PROSITE preprofiles |
N/A |
131 |
| ProDom |
2002.1 |
1.021 |
| InterPro |
7.1 |
10.403 |
| Smart |
3.4 |
654 |
| TIGRFAMs |
3.0 |
1.977 |
| PIR Superfamily |
2.3 |
219 |
| SUPERFAMILY |
1.63 |
552 |
|
La versión de InterPro 7.1 contiene 10.403 entradas (2.239 dominios,
7.901 familias,
197 'repeats',
26 centros activos,
20 'binding sites'
y 20 sitios de modificación
post-transduccional).
Hay 5.211.420 conexiones entre las 1.152.185 secuencias de Swiss-Prot
+ TrEMBL y las 10.403 entradas de InterPro.
El 93% de las proteínas de Swiss-Prot (y el 81% de las de TrEMBL)
tienen al menos una conexión con InterPro.
Lo que InterPro entiende por familias, dominios, repeticiones
('repeats') y modificaciones post-transduccionales
Familia: es un grupo de proteínas relacionadas evolutivamente
y que tienen uno o más dominios o repeticiones en común.
Una familia puede contener un 'motivo' que la defina.
Dominio: un dominio es una unidad estructural independiente. Una
entrada de InterPro del tipo dominio se puede usar como diagnóstico
de la presencia de un dominio, pero no tiene por qué definir correctamente
los límites del dominio.
Repetición: es una corta región que no tiene entidad
estructural independiente, es decir, se requiere la presencia de varias
repeticiones para dar lugar a un dominio. Ejemplo: el dominio WD40 está
constituido por 6 ú 8 copias de la repetición WD40.
Modificación post-transduccional (PTM): se refiere a motivos
de secuencia que son reconocidos en la célula para que se produzcan
PTMs sobre la proteína, como por ejemplo N-glicosilaciones, farnesilaciones,
etc... Las proteínas que son agrupadas de acuerdo a un PTM no tienen
por qué compartir un origen evolutivo.
Sitios de unión: son sitios responsables de la unión
de ligandos que por sí mismos no son sustratos de reacción
enzimática.
Centros activos: son sitios de unión en los que se produce
una reacción enzimática.
Las familias se pueden dividir según relaciones de 'padres'
e 'hijos', en familias y subfamilias (PARENT/CHILD relationships).
Por otra parte, en las entradas de dominios, hay un campo de
'contains/found in' para indicar si alguna familia ha sido caracterizada
por este dominio.
|
Ejemplo: el dominio CARD.
En su entrada se indica que hay 135 proteínas que tienen este dominio.
También se indica que los dominios correspondientes en Pfam, Prosite
y Smart (PF00619, PS50209 y SM00114) contienen 88, 133 y 76 proteínas,
respectivamente.
También se muestra una descripción de la función
de este dominio.
En el enlace "overview"
(y en "...sorted by name" y en "detailed")
se muestra gráficamente en qué regiones de cada una de las
135 proteínas aparece cada uno de los dominios Pfam, Prosite y Smart.
También se muestra qué otros dominios presentan cada una
de las 135 proteínas. Vemos que entre esas proteínas
algunas presentan el dominio "proteína kinasa", otras el dominio
"caspasa", etcétera. En "detailed" se muestra lo mismo pero más
ampliamente. Por ejemplo, en la parte de "overview" vemos esto para la
proteína RIK2_HUMAN:

Vemos que la proteína PIAP_PIG tiene varios dominios. Los dos
primeros (los azules) son "BIR repeats" (si ponemos el ratón encima
aparece una etiqueta que lo indica), luego está el dominio CARD
y luego el dominio Zn-finger de tipo RING.
Y en "detailed" vemos esto:

Vemos que en el caso de la proteína PIAP_PIG el dominio CARD
se encuentra con los tres marcadores: Pfam, Prosite y Smart (no ocurre
lo mismo con todas las proteínas que según interPro tienen
este dominio). Las repeticiones BIR
se encuentran con 4 marcadores (PF0065, PS01282, PS50143 y SM00238). Los
dominios
Zn-finger
se encuentran con tres marcadores. Al lado de cada marcador vemos con qué
entrada de InterPro se corresponde.
En el enlace "table",
vemos qué regiones de cada una de las 117 proteínas se corresponden
con cada una de las "signatures".
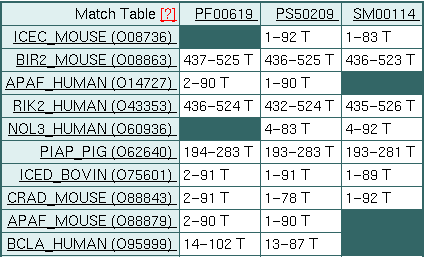
En la tabla además vemos cuáles de las "signatures" (marcadores)
aparecen en cada proteína. En PIAP_PIG aparecen las tres, pero en
NOL3_HUMAN, por ejemplo, no aparece el de Pfam. También se indica
mediante las letras "T", "F" y "?", si la relación es correcta (T,
true),
incorrecta (F, false) o no se sabe (?).
El caso de PIAP_PIG: ¿qué función podría
tener?. Esta proteína está anotada en Swiss-Prot
como un posible inhibidor de la apoptosis. El dominio CARD es un dominio
de interacción entre proteínas. Normalmente interacciona
con otros dominios CARD, los cuales están presentes en caspasas,
kinasas, etc. (las caspasas son las principales ejecutoras del plan apoptótico
de la célula). Por otra parte, el dominio BIR (que está constituido
por varias repeticiones BIR) confiere resistencia a apoptosis. Además,
BIR normalmente aparece asociado al dominio Zn-RING finger, el cual está
implicado en interacciones proteína-proteína.
De forma rápida hemos podido hacernos una idea de la función
de esta proteína y de qué dominios tiene y qué rol
puede que desempeñen. Posiblemente, de confirmarse que esta proteína
es inhibidora de la apoptosis, lo haga interfiriendo en la maquinaria apoptótica,
quizás uniéndose a ella a través del dominio CARD,
y usando el Zn-RING finger para alguna otra interacción. Este tipo
de dedos de Zinc están asociados a muchos procesos celulares, como
por ejemplo a la ubiquitinación (etiquetado de proteínas
para su destrucción), ya que puede interaccionar con enzimas ubiquitinadoras,
pero no sabemos si éste es el caso.
Clasificación jerárquica en
InterPro
Por ejemplo, para las proteínas kinasas existe esta jerarquía:

P.e. el IPR00719 (el nodo superior) está
definido por un dominio de ProDom (PD000001), también por uno de
Pfam (PF00069) y por otros dos: un patrón y un perfil de Prosite
(PS00107 y PS50011).
P.e. el nodo de las "Tyrosine protein kinases", el IPR001245,
está definido por una entrada de Prints (PR00109), otra de Prosite
(PS00109) y otra de SMART (SM00219).
etcétera.
COGs: Clusters of
Orthologous Groups (grupos de ortólogos)
[http://www.ncbi.nlm.nih.gov/COG/]
La documentación de COGs se encuentra en este
enlace.
COGs se refiere a "clusters of orthologous groups", es decir a grupos
de genes ortólogos. Su objetivo es clasificar en tales grupos
las proteínas de aquellos microorganismos de los que se conoce el
genoma al completo. En la última versión había 43
genomas.
En este momento están diseñando una nueva versión
de COGs que también será extensible a organismos pluricelulares
como Arabidopsis thaliana, Caenorhabditis elegans,
Drosophila
melanogaster o Homo sapiens.
Clasificar las proteínas en grupos de ortólogos es muy
útil para predecir la función de las mismas, ya que
ésta se suele conservar en los ortólogos y entonces, conociendo
la función de al menos una de las proteínas del grupo, podemos
saber cuál es la función de las otras.
Además, es útil para comparar genomas, ya que para
comparar el contenido génico de los genomas o la organización
de sus genes, también es necesario conocer las relaciones de ortología.
Identifiación de ortólogos basada en best bidirectional
hits (BBHs)
La idea del método de 'best bidireccional
hits (BBHs)' o mayores parecidos en las dos direcciones es
que si una proteína de un genoma es la más parecida de otra
en otro genoma, y viceversa (bidireccional), entonces es muy probable que
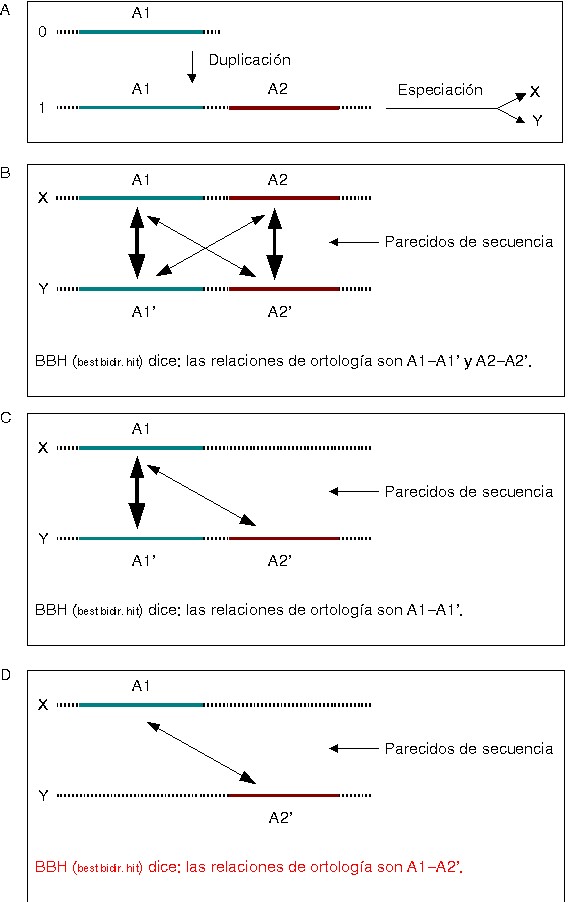
ambas sean ortólogas. Por ejemplo, supongamos que tenemos dos genomas
X e Y, y en cada uno de ellos hay dos proteínas homólogas
A1 y A2 que proceden de una duplicación ancestral en un genoma 0
(A en la figura). Si ninguno de los dos genomas X e Y sufre una
deleción, el método funcionará correctamente (caso
B).
Si se produce una deleción, dependiendo de a qué genes afecte,
el método funcionará bien (C) o mal (D).
|
El método de determinación de grupos de ortólogos
de COGs:
Es un método semiautomático, después de
aplicar el método que a continuación se describe, se realiza
una corrección de los resultados.
-
Lo primero que se hace es determinar los BeTs (best bidirectional hits):
para cada proteína se determina cuál es la más parecida
en cada uno de los otros genomas.
-
Fusión de in-paralogs: Como los in-paralogs (las duplicaciones
recientes dentro de una misma especie) pueden crear confusión (puede
que no tenga sentido determinar con cuál de los in-paralogs se ha
de establecer el BBH; puede que sea imposible determinarlo), lo que se
hace es fusionarlos, tomándolos como si fueran uno solo.
El criterio para la fusión es que su parecido sea más elevado
entre ellos que con respecto a cualquier otro gen de otro genoma.
-
Con las relaciones de BeTs se construye un grafo a partir del cual se buscan
patrones consistentes de BeTs. El más sencillo de éstos es
un triángulo de genes procedentes de tres linajes diferentes.
-
Los triángulos que comparten dos vértices (un lado) se unen.
Este procedimiento supuestamente resulta en grupos de ortólogos.
-
En muchos casos, dos o más grupos de ortólogos quedan unidos.
En esos casos se construye un árbol filogenético y se separan
manualmente.
Finalmente, se asigna una función particular a cada COG y también
una clase
funcional general.
|
El sitio web de COGs
ofrece muchas herramientas para consultar los resultados.
Se pueden buscar COGs por "palabras
clave", por representación
filogenética (p.e. obtener todos aquellos COGs en los que hay
arqueas pero no bacterias), por clase
funcional, por ruta
metabólica, etcétera.
Una vez identificado un COG de interés (ejemplo)
podemos ver qué función tiene o qué genomas están
representados en él.
-
Si pinchamos en cualquiera de las proteínas aparecerá una
lista de a qué proteínas se parece (ejemplo).
-
También se muestra un árbol filogenético. Sin pinchamos
en el árbol
veremos un análisis de componentes principales.
-
Si pinchamos en "Genome
context" podremos ver la organización de los genomas representados
en ese COG respecto al gen de interés. Una conservación en
la organización genómica puede ayudarnos a predecir la función
de genes desconocidos: si dos genes suelen aparecer juntos, incluso en
genomas distantes, posiblemente exista una relación funcional entre
ellos. Echad un vistazo al contexto genómico en del ejemplo
y pensad qué otros COGs aparecen en sus alrededores. ¿Tienen
relación funcional? ¿Por qué creéis que aparecen
juntos en organismos tan distantes?. (Respuesta: tiene que ver con la organización
en operones en las bacterias).
-
También podemos buscar qué otros COGs tienen un mismo patrón
filogenético, lo cual puede indicar la existencia de interacciones
funcionales entre ambos genes.
En COGs también existen herramientas para comparar genomas.
Por ejemplo viendo su organización estructural, como en el ejemplo
anterior. También hace un análisis
de componentes principales basado en la co-ocurrencia
de los genomas en los COGs.
Por otra parte, si queremos clasificar una secuencia podemos utilizar
el programa COGNITOR
(ayuda).
Podemos buscar con swiss:DNAK_ECOLI.
>DNAK_ECOLI|P04475|Chaperone protein dnaK (Heat shock protein
70) (Heat shock 70 kDa protein) (HSP70).
GKIIGIDLGT TNSCVAIMDG TTPRVLENAE GDRTTPSIIA
YTQDGETLVG QPAKRQAVTN
PQNTLFAIKR LIGRRFQDEE VQRDVSIMPF KIIAADNGDA
WVEVKGQKMA PPQISAEVLK
KMKKTAEDYL GEPVTEAVIT VPAYFNDAQR QATKDAGRIA
GLEVKRIINE PTAAALAYGL
DKGTGNRTIA VYDLGGGTFD ISIIEIDEVD GEKTFEVLAT
NGDTHLGGED FDSRLINYLV
EEFKKDQGID LRNDPLAMQR LKEAAEKAKI ELSSAQQTDV
NLPYITADAT GPKHMNIKVT
RAKLESLVED LVNRSIEPLK VALQDAGLSV SDIDDVILVG
GQTRMPMVQK KVAEFFGKEP
RKDVNPDEAV AIGAAVQGGV LTGDVKDVLL LDVTPLSLGI
ETMGGVMTTL IAKNTTIPTK
HSQVFSTAED NQSAVTIHVL QGERKRAADN KSLGQFNLDG
INPAPRGMPQ IEVTFDIDAD
GILHVSAKDK NSGKEQKITI KASSGLNEDE IQKMVRDAEA
NAEADRKFEE LVQTRNQGDH
LLHSTRKQVE EAGDKLPADD KTAIESALTA LETALKGEDK
AAIEAKMQEL AQVSQKLMEI
AQQQHAQQQT AGADASANNA KDDDVVDAEF EEVKDKK
ProtoMap
[http://protomap.cornell.edu/]
Una introducción
y un tour por
ProtoMap.
| ProtoMap es una base de datos que ofrece una clasificación
jerárquica del espacio de secuencias. El método es totalmente
automático.
La clasificación se realiza en función de las distancias
entre las secuencias, en función de cuánto se parecen.
Se realiza una búsqueda mediante Smith & Waterman, BLAST
y FASTA para cada una de las secuencias de Swiss-Prot+TrEMBL.
De este modo se obtiene una medida de la distancia entre todas las
proteínas.
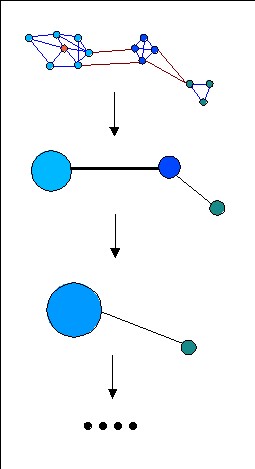
Estos resultados se representan mediante un grafo en el que los
nodos
son las proteínas y los nodos están unidos por
arcos
cuando alguno de los métodos ha encontrado un parecido. El peso
de los arcos viene dado por el e-value asociado a tal parecido de secuencia.
En este grafo las proteínas que se parezcan más estarán
más cerca entre sí (ejemplo). El
objetivo ahora es encontrar un algoritmo de clustering (agrupamiento)
capaz de identificar automáticamente los grupos de secuencias que
existen en dicho mapa.
El algoritmo de clustering:
El algoritmo funciona de forma recursiva. Aplicando un umbral cada vez
más suave se van obteniendo grupos más grandes. Es equivalente
a ir uniendo subfamilias en familias y éstas en superfamilias.
0º.- Obtención de distancias entre secuencias =>
grafo
1º.- Agrupamiento de secuencias claramente relacionadas (e-value
< 1e-100)
2º.- Inicialización de T = 1e-95.
3º.- Cálculo de distancias entre los distintos grupos o
clusters:
-Se halla la media geométrica de los e-values entre
cada par de clusters. En los casos en que no hay arcos, se asigna un e-value
de 1.
4º.- Si la media geométrica de los e-values es menor que la
raíz cuadrada de T, se unen los clusters.
5º.- Se relaja el umbral T: T = T*1e+05.
6º.- Si T > 1 => FIN. Si no => se vuelve al punto 3º.
La aplicación de distintos T secuencialmente (1e-95 -> 1e-90
-> 1e-85 ... 1e-00=1) resulta en una clasificación jerárquica
de las proteínas.
|
|
|
Problemas de esta aproximación:
-la propia limitación de los métodos de comparación
entre pares de secuencia, que tienen un menor poder de discriminación
de homologías remotas. Sin embargo la comparación entre pares
de secuencias tiene la ventaja de que ofrece una medida de la distancia
evolutiva entre las secuencias (no así la comparación entre
un perfil y una secuencia) lo cual permite aplicar el método de
agrupamiento automático.
-la propiedad transitiva de la homología entre las proteínas
se aplica sin tener en cuenta la naturaleza multidominio de las mismas,
por lo que en un mismo cluster puede haber proteínas que no tengan
un mismo origen.
Interfaz web:
Se puede clasificar una nueva secuencia (realizando una búsqueda)
o bien se puede navegar por la jerarquía
de clusters, buscando por "keywords", "accession number", etc. Se puede
seleccionar también el nivel al que queremos acceder, desde 1e-0
hasta 1e-100, desde grupos más grandes a grupos más pequeños.
Ejemplo: swiss:RASH_HUMAN.
Buscando con el identificador de swiss-prot averiguamos que esta proteína,
en el nivel 1e-0, se encuentra en el cluster 18,
que contiene 976 proteínas. También vemos con qué
otros clusters tiene arcos el nodo de rash_human.
Si vamos al cluster 18
veremos qué proteínas hay en él, su función,
su origen filogenético (virus, eucariotas, bacterias, etc). También
vemos qué patrones y perfiles de Prosite están presentes
en las proteínas del grupo.
También podemos:
-
ver el alineamiento
mútliple.
-
ver los constituyentes de mayor orden ("higher
order constituens"), donde podemos ver a partir de qué subgrupos
se ha constituido este cluster (es decir, los grupos que había a
umbrales más restrictivos).
-
Y también podemos ver con qué otros clusters tienen relaciones
(arcos, parecidos de secuencia) las proteínas de este grupo ("possibly
related clusters"). Allí veremos los grupos vecinos ordenados
de acuerdo a la "quality" que no es más que la media geométrica
de los e-values, calculada como se ha mencionado antes. También
vemos cuántos arcos hay entre los grupos y el tamaño de los
grupos. Es muy útil para ver con qué otras familias está
relacionada una familia determinada.
-
En el servidor antiguo
de ProtoMap sólo se aplicaba el método sobre Swiss-Prot por
lo que se ha quedado un poco anticuado, pero tenía la ventaja de
que permitía consultar el origen del grupo en forma de árbol,
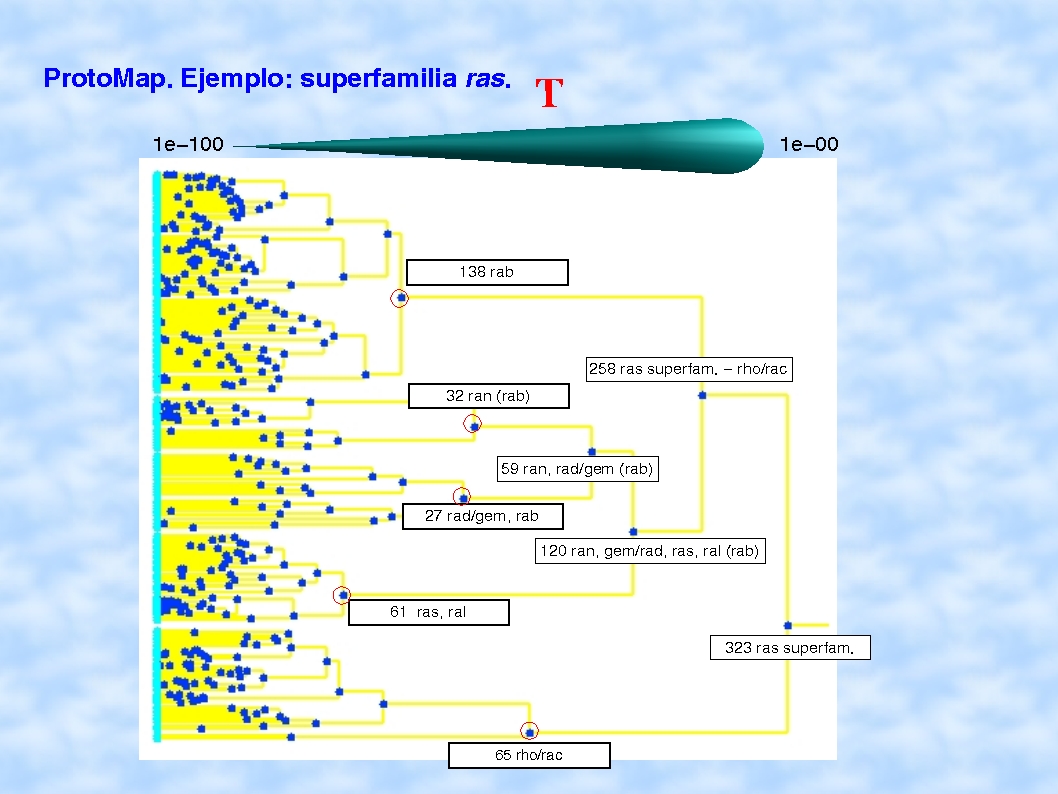
de forma que podíamos ver qué grupos se habían ido
uniendo según se relajaba el umbral y qué proteínas
había en cada uno, como en esta figura.
Cuestión práctica:
¿Hay
alguna relación evolutiva entre la proteína ras/p21 de humanos
y el factor de elongación de la traducción EF-Tu de E. coli?
Estas son sus secuencias:
>RASH_HUMAN|P01112|Transforming protein P21/H-RAS-1 (C-H-RAS).
MTEYKLVVVG AGGVGKSALT IQLIQNHFVD EYDPTIEDSY
RKQVVIDGET CLLDILDTAG
QEEYSAMRDQ YMRTGEGFLC VFAINNTKSF EDIHQYREQI
KRVKDSDDVP MVLVGNKCDL
AARTVESRQA QDLARSYGIP YIETSAKTRQ GVEDAFYTLV
REIRQHKLRK LNPPDESGPG
CMSCKCVLS
>EFTU_ECOLI|P02990|Elongation factor Tu (EF-Tu) (P-43).
SKEKFERTKP HVNVGTIGHV DHGKTTLTAA ITTVLAKTYG
GAARAFDQID NAPEEKARGI
TINTSHVEYD TPTRHYAHVD CPGHADYVKN MITGAAQMDG
AILVVAATDG PMPQTREHIL
LGRQVGVPYI IVFLNKCDMV DDEELLELVE MEVRELLSQY
DFPGDDTPIV RGSALKALEG
DAEWEAKILE LAGFLDSYIP EPERAIDKPF LLPIEDVFSI
SGRGTVVTGR VERGIIKVGE
EVEIVGIKET QKSTCTGVEM FRKLLDEGRA GENVGVLLRG
IKREEIERGQ VLAKPGTIKP
HTKFESEVYI LSKDEGGRHT PFFKGYRPQF YFRTTDVTGT
IELPEGVEMV MPGDNIKMVV
TLIHPIAMDD GLRFAIREGG RTVGAGVVAK VLS
Si queréis podéis probar con BLAST
para determinar esta cuestión. Pero os adelanto que el mejor alineamiento
local que obtendréis será muy pequeño y poco significativo
(=no distinguible de un parecido al azar entre el millón de proteínas
conocidas):
ras : ESRQAQDLARSYGIPYI
eftu: QTREHILLGRQVGVPYI
¿Podríamos haber detectado la relación evolutiva
entre rash_human y EF-Tu de ecoli usando Pfam?
Los perfiles y
los HMMs permiten determinar relaciones evolutivas distantes porque incorporan
información precisa de la familia en cuestión, como por ejemplo
que los residuos X e Y sean un Trp y una Lys, dando más importancia
a la conservación de éstos que a la de otros.
1º.
Id a Pfam y pinchad
en el enlace de 'protein
search'.
2º.
Pegad la secuencia de rash_human y poned el 'E-value
cutoff level' a 100. Obtendréis esto.
MTEYKLVVVG AGGVGKSALT IQLIQNHFVD EYDPTIEDSY
RKQVVIDGET CLLDILDTAG
QEEYSAMRDQ YMRTGEGFLC VFAINNTKSF EDIHQYREQI KRVKDSDDVP
MVLVGNKCDL
AARTVESRQA QDLARSYGIP YIETSAKTRQ GVEDAFYTLV REIRQHKLRK
LNPPDESGPG
CMSCKCVLS
3º. Haced lo mismo con eftu_ecoli en otra ventana. Obtendréis
esto.
SKEKFERTKP HVNVGTIGHV
DHGKTTLTAA ITTVLAKTYG GAARAFDQID NAPEEKARGI
TINTSHVEYD TPTRHYAHVD CPGHADYVKN MITGAAQMDG AILVVAATDG
PMPQTREHIL
LGRQVGVPYI IVFLNKCDMV DDEELLELVE MEVRELLSQY DFPGDDTPIV
RGSALKALEG
DAEWEAKILE LAGFLDSYIP EPERAIDKPF LLPIEDVFSI SGRGTVVTGR
VERGIIKVGE
EVEIVGIKET QKSTCTGVEM FRKLLDEGRA GENVGVLLRG IKREEIERGQ
VLAKPGTIKP
HTKFESEVYI LSKDEGGRHT PFFKGYRPQF YFRTTDVTGT IELPEGVEMV
MPGDNIKMVV
TLIHPIAMDD GLRFAIREGG RTVGAGVVAK VLS
¿Qué conclusión podéis sacar? ¿Es capaz
de detectar la relación evolutiva? ¿Sale el dominio característico
de cada una de estas proteínas en la lista de 'matches' del otro?
¿Con qué E-value? ¿Son iguales estos E-values? ¿Por
qué?
{kind=link}
{kind=link}