
http://www.rcsb.org/pdb

H. influenzae
Las características 1D de una secuencia son aquellas que pueden ser representadas por un solo valor asociado a cada aminoácido (B. Rost). Entre ellas incluiríamos los valores de características fisico-químicas de amino-ácidos, o las predicciones de estructura secundaria.
Las características 2D de las proteínas, por otro lado, corresponden a la descripción de los contactos entre los residuos de la proteína, ya sea a corta distancia o a larga distancia. Por contactos se entiende cualquier tipo de enlace (puente de hidrogeno, puente disulfuro), entre residuos. Los contáctos a corta distancia están relacionados con el tipo de estructura secundaria. Los contactos a larga distancia, sin embargo, dan información de la organización de los elementos de estructura secundaria. La prediccion de contactos, como herramienta para predecir la estructura terciaria es un campo poco desarrollado aún.
Las características 3D se refieren a la estructura terciaria de las proteínas.
Como consecuencia de los proyectos de secuenciación de genomas iniciados en los últimos años se ha incrementado el volumen de las bases de datos de secuencias (SWISS-PROT) aunque no así el de bases de datos de estructuras (PDB).
Actualmente no existen métodos capaces de
a partir de su secuencia predecir la estructura 3D de una proteína.
Sin embargo, si que se dispone de métodos capaces de predecir aspectos
mas sencillos de su estructura, esto es, las características 1D,
a partir de los cuales se puede derivar cierta información sobre
su posible función y su estructura.
|
http://www.rcsb.org/pdb |
H. influenzae |
Se ha hipotetizado, y ha sido verificado para muchas proteínas, que la estructura 3D de una proteína (esto es su plegamiento) viene determinada únicamente por la especificidad de la secuencia. Por otro lado se sabe que las proteínas chaperonas juegan a menudo un papel fundamental en el plegamiento, y aun así se asume generalmente que la estructura final es la que representa el mínimo de energía libre. Es por esto que se afirma que toda la información sobre la estructura nativa de una proteína esta codificada en su secuencia amino acídica, aunque es especifica del medio en solución en que se encuentre. Sin embargo en la practica, la falta de precisión en determinación de los parámetros básicos de los que se derivaría la estructura 3D y los recursos informáticos limitados hacen que los métodos predicción mas fiables sean aquellos basados en el conocimiento, combinación de métodos estadísticos y empíricos. Sin embargo, como se ha demostrado en los experimentos de CASP (de los que os hablaran en próximas lecturas) no se puede aun predecir estructura a partir de secuencia.
Una simplificación del problema de predicción
de estructura 3D es su proyección en cadenas de asignaciones estructurales.
Por ejemplo, podemos asignar estados de estructura secundaria o solvatación
para cada residuo identificándolos con un símbolo. De hecho,
los mayores avances en bioinformática de la ultima década
se han alcanzado en el campo de la de predicción de estructura secundaria.
Estos avances se han alcanzado al combinar de algoritmos matemáticos
complejos con la información evolutiva disponible en las bases de
datos.
 |
Las predicciones de características 1D, aunque locales e incluso a veces parcialmente correctas, son a menudo útiles para obtener información sobre la función de la proteína y/o los sitios activos y para llevar a cabo predicciones de mas complejas (aspectos estructurales de mayor dimensión).
Propiedades de los residuos
La información mas inmediata que podemos sacar de la secuencia de una proteína son características físico-químicas de sus residuos: hidrofobicidad, polaridad, etc. Con esto podemos generar representaciones de, por ejemplo, como varía la hidrofobicidad a lo largo de la secuencia de la proteína para tener información como zonas muy hidrofóbicas, etc que puedan luego ayudarnos en la predicción de características estructurales.
Hay muchas herramientas que calculan este tipo de parámetros a partir de la secuencia. Muchas han sido dotadas de interfaces WWW que facilitan en gran medida su uso.
Este servidor toma como input una secuencia y permite elegir entre 54 parámetros.
Como output devuelve una representación gráfica de como varia ese parámetro a lo largo de la secuencia y un fichero con esos datos en forma numérica que otros programas pueden leer.

Perfil de Hidrofobicidad Kyte-Doolitle
Accesibilidad
El objetivo es la predicción de la exposición de un residuo al solvente. La accesibilidad se puede describir de varias formas.
El método clásico asignaba uno de los valores "buried/exposed" en función de la hidrofobicidad del residuo. Con este método zonas muy hidrofóbicas son predecidas como "buried". El resultado de éste tipo de predicción se correlaciona, por tanto, con la predicción del perfil de Hidrofobicidad.
Sin embargo, métodos mas avanzados emplean análisis similares a los que se emplean en la predicción de estructura secundaria (que se comentan más adelante), como por ejemplo, el uso de redes neuronales u otros algoritmos que se entrenan con proteínas de estructura conocida, para las que se puede calcular la accesibilidad real de cada residuo al solvente.
Cuando la estructura de la proteína es conocida,
la accesibilidad de cada residuo se calcula estimando el volumen expuesto
al solvente de cada residuo embebido en una estructura (método desarrollado
por Connolly y implementado posteriormente en DSSP). Una simplificación
del mismo seria pasar de los valores normalizados (el valor observado dividido
por el máximo valor posible) a una descripción con dos posibles
estados "buried" (accesibilidad relativa < 16%) y "exposed" (accesibilidad
relativa
> 16%),tal y como se hacía con los métodos clásicos
basados en hidrofobicidad.

Dickerson´s Dodecamer |
 |
Medida de la accesibilidad al solvente (Fig. from B. Rost). La accesibilidad al solvente es normalmente medida haciendo rodar una molécula de agua esférica sobre la superficie de la proteína, el valor total es la suma de la superficie correspondiente a cada uno de los residuos (normalmente 0-300 Å2). Con objeto de comparar aminoácidos se calculan valores relativos (esto es el porcentaje de área accesible). Descripciones mas sencillas diferencian tan solo entre dos estados: buried (en la figura residuos 1-3 y 10-12) y exposed (residuos 4-9). |
La accesibilidad para cada posición de la estructura 3D de la proteína es evolutivamente conservada dentro de cada familia de secuencias. Por lo tanto la información contenida en los alineamientos de secuencias se ha empleado para desarrollar de nuevos métodos de predicción, pasando de una fiabilidad del 75% al 79%.
Entre los servidores públicos para calcular la accesibilidad a partir de la secuencia de aminoácidos, se encuentran:
JPred2. JPred2 emplea perfiles de PsiBlast como input para sus redes neuronales y devuelve dos estados "buried/exposed". JPred2 es una versión avanzada de Jpred.
Estructura secundaria
La predicción de la estructura secundaria de una proteína, partiendo de su secuencia de aminoácidos, busca definir los segmentos que adoptan una de los tres "ESTADOS" de estructura secundaria: alfa hélice, beta (o conformación extendida) y loop (o coil).
La mayoría de los métodos usan redes neuronales u otros algoritmos que se entrenan con proteínas de estructura secundaria conocida para pasar luego a la predicción. Muchos de estos métodos usan información adicional proveniente, por ejemplo, de alineamientos múltiples.
La estructura secundaria de las proteínas
usadas en la fase de "entrenamiento" (durante el desarrollo del programa)
se asigna generalmente de forma automática, una vez se conoce la
estructura terciaria de la proteína, en función de su perfil
de puentes de hidrogeno entre los grupos carbonilos y NH del esqueleto
o "backbone". El programa que más frecuentemente se usa para hacer
esa asignación es DSSP.
Precedentes históricos de los métodos de predicción de estructura secundaria:
1957 Szent-Györgyi & Cohen intentan correlacionar el contenido de ciertos aminoácidos, tales como la prolina, con el contenido en hélices alfa de una secuencia proteica.
1960 Blout, Fasman et al. & 1962 Blout, amplían esta idea en el sentido de correlacionar el contenido total de aminoácidos con el de hélices alfa y láminas beta.
1960. Kendrew et al. & Perutz et al, determinan experimentalmente mediante espectrometría de rayos-X las primeras estructuras de proteínas: mioglobina y hemoglobina.
1978. Garnier mejoro el método al emplear pares de interacciones significativas estadísticamente. El método presenta una fiabilidad de ~60%.
1993. Levin mejoro las predicciones empleando alineamientos múltiples de secuencia. Regiones conservadas dentro del alineamiento proporcionan un fuerte indicador evolutivo de su importancia en la función. Estas zonas conservadas tienen además tendencia a conservar su estructura, reforzando la predicción. El método presenta una fiabilidad de ~69%.
1994. Rost y Sander combinaron redes neuronales con alineamientos múltiples de secuencias. El método presenta una fiabilidad de ~72%.
Estos son métodos estadísticos basados en la tendencia que presentan los aminoácidos a adoptar estructuras secundarias.El primero, propuesto por Chou y Fasman en 1974 empleaba estadísticas extrapoladas de las 15 estructuras de proteínas determinadas por rayos-X.
Tendencias que se basaban en las propiedades estereoquímicas y fisicoquímicas de los diferentes residuos (casos especiales son glicina y prolina). Este método se ha mejorado aumentando el número de proteínas empleadas.
El método presenta una fiabilidad de ~50% (cuando se emplean 62 proteínas para obtener las estadísticas).
La principal mejora de esta 2a generación de métodos es la combinación de bases de datos mayores de estructura de proteínas y el uso de estadísticas basadas en segmentos: típicamente 11-21 residuos adyacentes y las estadísticas se compilan para evaluar la propensión del residuo central de ese segmento a estar en una determinada estructura secundaria.Los métodos de 1a y 2a generación presentaban problemas obvious:
fiabilidad (predicciones 3-estados) <70%
Esto es consecuencia de:fiabilidad para las betas 28-48% (~random)
alfas y betas demasiado cortas
las estructuras obtenidas experimentalmente difieren incluso de un cristal a otro
la estructura secundaria depende de interacciones de largo rango (a más de 11-21 residuos adyacentes). Este hecho se acentúa más para betas que alfas.
La incorporación de la información evolutiva permite una mejora de estas predicciones. Los perfiles de intercambio de residuos extraídos de los alineamientos de una familia son indicativos de detalles estructurales específicos. Además estos perfiles implícitamente contienen información no local, ya que la selección evolutiva de proteínas se hace a nivel de estructura 3D y no a nivel de secuencia.Los perfiles extendidos conseguidos a través de PsiBlast y Hidden-Markov-Models mejoran por tanto las predicciones.
Ventajas:
fiabilidad (predicciones 3-estados) 70%
Problemas:fiabilidad para las betas ~ alfa ~ "loops"
malos alineamientos llevan a malas predicciones
confusión de alfas y betas se de en regiones en que se establecen interacciones a largo rango
precaución al evaluar los resultados para proteínas con características inusuales
| Kyte-Doolittle Hydropathy Scale
|
Scheme for PHD Protein Predictor
Methods
|

Ejemplo de la salida de tres servidores de predicción de estructura secundaria. La secuencia pertenece a un dominio SH3. La estructura secundaria observada fue asignada con DSSP. Los niveles de fiabilidad de las predicciones son: C+F = 59%, GORIII = 65% y PHD = 72%. El "reliability index" presenta valores de 0-9. Para valores de Rel > 4 la predicción fue correcta. |

Ejemplo de la fiabilidad (3-estados/residuo) de diferentes servidores de predicción de estructura secundaria. Métodos de 1a generación: Chou & Fasman, Lim, GORI Métodos de 2a generación: Schneider, ALB, GORIII Métodos de 3a generación: LPAG, COMBINE, S83, NSSP, PHD |
Los servidores públicos disponibles son:
JPred y Jpred2 son métodos que incorporan redes neuronales e incluye información evolutiva (PsiBlast). La versión 2 evalúa los resultados de 4 redes neuronales diferentes (JNet, NSSP, Predator, PHD) para mejorar la fiabilidad.
PROF, no relacionado con PROFsec (más abajo aparece como PROF-King, por ser éste uno de los autores del programa), está basado en alineamientos múltiples y otras características de los residuos obtenidas de bases de datos. Fiabilidad de ~70%.
PSIpred Basado en perfiles generados con psi-blast (filtrando los resultados eliminando proteínas extrañas) y redes neuronales (combina los resultados de varios métodos de predicción de estructura secundaria). Acierto >76%.
SAM-T99 Una red neuronal y perfiles de alineamientos múltiples mejorados mediante el empleo de "Hidden Markov".
SSpro Emplea redes neuronales recurrentes y bidireccionales de ventanas fijas y de pequeño tamaño que permiten usar la cadena proteica completa como input.
Hélices Transmembrana
En el campo de la proteómica uno de los mayores
retos es la determinación de la estructura de proteínas transmembrana,
ya que son difíciles de cristalizar y son difícilmente analizables
con NMR. Por lo tanto, la predicción de la estructura de este tipo
de proteínas es de un mayor interés. Existen dos clases principales
de proteínas de membrana : las que introducen hélices en
la bicapa lipídica (Figura) y, proteínas que forman poros
constituidos por barriles de betas (tipo porinas). Hasta el momento no
existen servidores públicos para este segundo grupo debido a la
falta de información experimental. La situación es muy diferente
para las hélice transmembrana. La estructura 3D se puede determinar
conociendo la precisa localización de las hélices transmembrana
explorando simplemente todas las conformaciones posibles.

| Hélices de membrana (Fig. from B. Rost). Para cierta clase de proteínas de membrana, típicamente los segmentos apolares de la hélice se encuentran embebidos en la bicapa lipídica orientados de forma perpendicular a la superficie de la membrana. Las hélices pueden ser consideradas como cilindros rígidos. La orientación de las hélices con respecto a la célula puede ser definida por la orientación del primer residuo N-terminal. La topología es definida como fuera cuando el N-terminal esta en la región extra-citoplasmática (proteína A), y como dentro si el N-terminal empieza en la región intra-citoplasmática (proteínas B y C). La parte inferior explica la regla de 'inside-out-rule'. |
A pesar de la dificultad de para su determinación experimental, estas proteínas presentan fuertes restricciones estructurales ya que la bicapa lipídica reduce los grados de libertad. Las hélices transmembrana se pueden predecir a partir de observaciones que limitan el problema: (a) estas hélices son predominantemente apolares y con una longitud de 12-35 residuos, (b) las regiones globulares entre hélices presentan típicamente longitudes menores de 60 residuos, (c) la mayoría de las hélices transmembrana tienen una distribución característica de los aminoácidos positivos arginina y lisina (definida en la regla 'positive-inside-rule' by Gunnar von Heijne) de forma que los "loops" en la zona interior de la membrana tienen mas cargas positivas que los "loops" en la zona exterior de la misma, (d) las regiones globulares largas (> 60 residuos) difieren en su composición de aquellas sujetas a la regla 'positive-inside-rule'.
La mayoría de los métodos se basan en redes neuronales u otros algoritmos que se entrenan con proteínas de estructura conocida. Se consiguen mayores porcentajes de acierto ya que estas hélices suelen tener patrones muy claros (anfipaticidad, etc) que son rápidamente asimilados por los algoritmos de aprendizaje. La mayoría de los métodos compilan esta información de hidrofobicidad, y los mas avanzados incluyen la regla " positive-inside-rule" para predecir además la orientación en la membrana.La inclusión de información evolutiva mejora considerablemente las predicciones de hélices transmembrana, aunque esto se compensa de alguna forma con el crecimiento de las bases de datos de secuencias.
Los servidores públicos disponibles (listado):
MEMSAT introduce un programa dinámico de optimización para encontrar la mejor predicción basada en preferencias estadisticas.
TMAP emplea preferencias estadísticas y perfiles de alineamiento.
PHDthm/PROFthm combinan redes neuronales que emplean información evolutiva con programas dinámicos de optimización para mejorar la predicción.
DAS optimiza el uso de perfiles hidrofobicos.
SOSUI usa una combinación de preferencias hidrofóbicas y antipáticas para predecir hélices transmembrana.
TMHMM es el mas avanzado de los métodos y aparentemente el de mayor fiabilidad. Implementa la información estadística y estas reglas indicadas en los modelos matemáticos "Hidden Markov" para optimizar las predicciones y la localización y orientación de las hélices (un concepto similar es empleado por HMMTOP).

Hélices Transmembrana (Perfil TMHMM)
Predicción de Péptidos de Señal (SignalP)
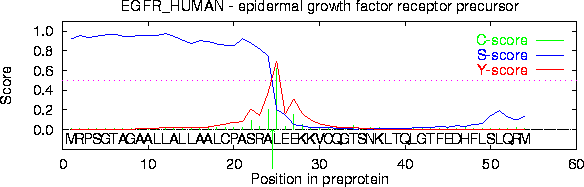
El servidor SignalP predice la presencia y localización de sitios de ruptura de péptidos señal en secuencias proteicas de diferentes organismos.
Dado que las características de los péptidos señales están relativamente bien conservadas y bién caracterizadas, es posible hacer predicciones con alta fiabilidad.
En las siguientes se muestran, respectivamente, las
características generales de los péptidos señales
en Eucariotas y en Procarioras Gram(+) y Gram(-), la distribución
de longitudes de péptido señal en esos tres grupos de organismos,
y un ejemplo de la salida de SignalP.


Modificaciones Post-transcripcionales
El servidor ExPASy Proteomics tools incluye una serie de herramientas para predecir características 1D.
TargetP predicción de la localización subcelular
SignalP predicción de péptidos señales
ChloroP predicción de péptidos de cloroplastos
MITOPROT predicción de secuencias diana de mitocondria
Predotar predicción de secuencias diana de mitocondria y plastidos
NetOGlyc predicción de sitios de O-glicosilacion en proteínas de mamíferos
NDictyOGlic predicción de sitos de GlcNAc O-glicosilación en "Dictyostelium"
YinOYang predicción de sitios de unión de O-beta-GlcNAc en proteínas eucariotas
big-PI Predictor predicción de sitios de modificación GPI (glicosil-fosfatidil inositol)
DGPI predicción de sitos de anclaje y ruptura de GPI
NetPhos predicción de sitios de fosforilación (Ser, Thr, Tyr) en eucariotas
NetPicoRNA predicción de sitos de ruptura para proteasas en proteínas de picornavirus
NMT predicción de sitos de N-miristoilación N-terminal
Sulfinator predicción de sitios de sulfatación de tirosina
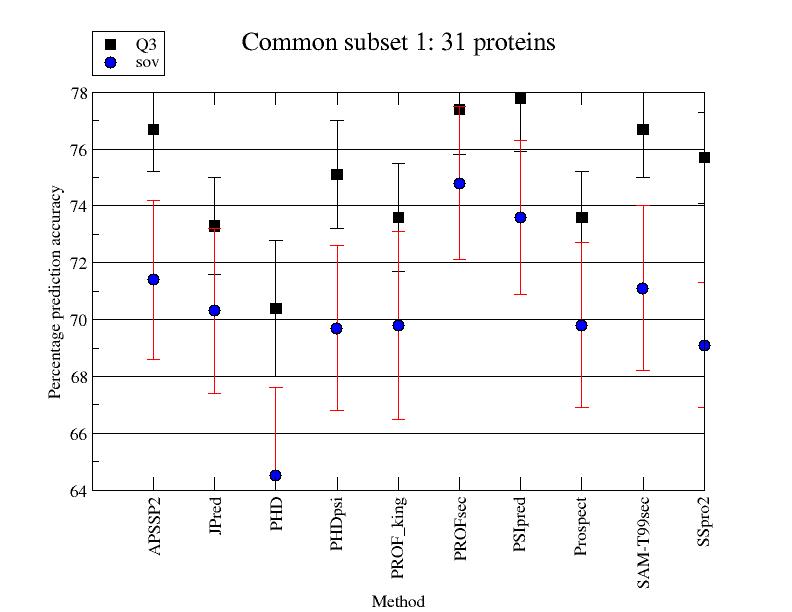
Evaluación automática de los servidores de predicción: EVA
EVA
es un sistema para la evaluación automática de servidores
de predicción de varios tipos:
Predicción de la función de proteínas a partir de características 1D
Como ya se ha adelantado más arriba, las características 1D de proteínas, además de proporcionar información que puede ser útil para predecir la estructura erciaria de proteínas, pueden ser muy útiles para predecir la funci'on de una proteína.
Un ejemplo muy obvio sería la detección de motivos característicos de un determinado tipo de enzima.
ProtFun
es un servidor que, utilizando información muy elemental, que en
principio no depende de la existencia de similaridades significativas en
las bases de datos, predice función a un nivel básico. Para
ello, calcula unos 20 parámetros de la proteína y usa una
red neuronal entrenada con información sobre proteínas de
funcion conocida.
volver


