Present and future uses of competition binding assays
It is too expensive and too technically challenging to characterize receptors only by direct radioligand binding assays. Moreover, when studying a new drug, it is usually not feasible to radiolabel the drug prior to understanding its receptor properties. For these reasons, competition assays are used most widely in receptor studies. One of the interesting new approaches in drug design and development is an elegant brute force approach called combinatorial chemistry. In this approach, one starts with several structural backbones that are felt likely to be components of a receptor pharmacophore (the structural features of a drug that cause binding and/or activation or blockade) and does chemical reactions that allow several different "pieces" to react in a way that hundreds of products may be formed. Rather than separating this gemisch and testing each component, the mixture is tested, and if the mixture is found to be "positive", the active species are purified and studied more conventionally. Such an approach dramatically increases the throughput of tested compounds. The most commonly used test system in combinatorial chemistry is the radioreceptor assay!
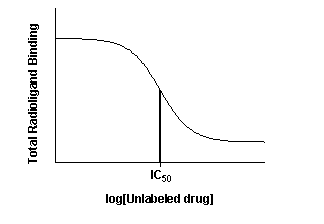
Competitive binding experiments measure the binding of a single concentration of labeled ligand in the presence of various concentrations of unlabeled ligand. Typically, the concentration of unlabeled ligand varies over at least six orders of magnitude.
The top of the curve is a plateau at a value equal to radioligand binding in the absence of the competing unlabeled drug. The bottom of the curve is a plateau equal to nonspecific binding. The concentration of unlabeled drug that produces radioligand binding half way between the upper and lower plateaus is called the IC50 (inhibitory concentration 50%) or EC50 (effective concentration 50%).
If the radioligand and competitor both bind reversibly to a single binding site, binding at equilibrium follows this equation (where Top and Bottom are the Y values at the top and bottom plateau of the curve).
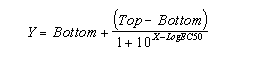
Competitive binding experiments are often used in more complicated situations where this equation doesn't apply.
If you performed an experiment with triplicate samples and you are using software for analysis, let the software automatically plot the error bars. Some investigators transform their data to percent specific binding. The problem with this approach is that you need to define how many cpm equal 100% binding and how many equal 0% specific binding. Deciding on these values is usually somewhat arbitrary. It is usually better to enter the data as cpm.
When analyzing the data, you need to decide whether to minimize the sum of the square of the absolute distances of the points from the curve or to minimize the sum of the square of the relative distances. The choice depends on the source of the experimental error. Follow these guidelines:
With experience, you will notice that even with the use of the best software, data that is non-ideal (i.e., most data) requires the experimenter to make decisions during the course of the analysis. Many times, your data will span a wide range of concentrations of unlabeled drug, and clearly define the bottom or top plateaus of the curve. If this is the case, software can estimate with accuracy the 0% and 100% values from the plateaus of the curve.
In some cases, your competition data may not define a clear bottom plateau, but you can define the plateau from other data. All drugs that bind to the same receptor should compete all specific radioligand binding and reach the same bottom plateau value. This means that you can define the 0% value (the bottom plateau of the curve) by measuring radioligand binding in the presence of a standard drug at a concentration known to block all specific binding. If you do this, make sure that you use plenty of replicates to determine this value accurately. If your definition of the lower plateau is wrong, the values for the EC50 will be wrong as well. You can also define the top plateau as binding in the absence of any competitor.
To summarize, if you have collected enough data to clearly define the entire curve, let the software fit all the variables and fit the top and bottom plateaus based on the overall shape of your data. If your data do not define a clear top or bottom plateau, you should define one or both of these values to be constants fixed to values determined from other data. Because of factors such as intra-assay drift, this often provides a challenge to the investigator. In the class exercises, change the top and bottom values, and observe the effects on the derived variables and the fitted curve.
To interpret the results, you must make these assumptions:
The IC50 is influenced by three theoretical factors (in addition to the factors controlled, or affected, by the experimenter :
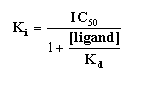
If the labeled and unlabeled ligand compete for a single binding site, the competitive binding curve will have a shape determined by the law of mass action. In this case, the curve will descend from 90% specific binding to 10% specific binding over an 81-fold increase in the concentration of the unlabeled drug. More simply, virtually the entire curve will cover two log units (100-fold change in concentration).
To quantify the steepness of a competitive binding curve, one determines a slope factor (also called Hill slope). A standard competitive binding curve that follows the law of mass action has a slope of -1.0. If the slope is more shallow, the slope factor will be a negative fraction (i.e. -0.85 or -0.60).
The Hill Equation provides information about the nature of ligand-receptor interactions. Widely used in the past, and often demanded by reviewers, its utility has been decreased by the wide availability of nonlinear regression analysis programs (like Prism, a program designed for receptor analysis). Thus, while less useful, it is important to understand the Hill equation since it has been a standard way of describing the slope factor (also called Hill slope or Hill number). It is derived from the binding equation modified to include the situation whereby the ligand may have multiple sites for interaction "n".
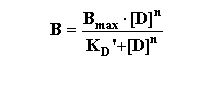
This can then be rearranged in several steps as follows:
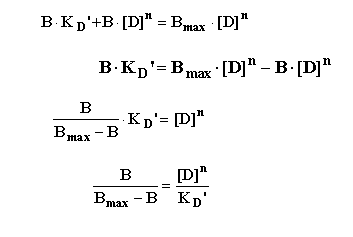
To yield the "Hill Equation".
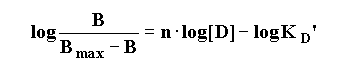
Thus, a plot of the log [Drug] vs. the logit function on the left hand side of the equation results in a straight-line with a slope = nH. (the Hill slope or Hill number)
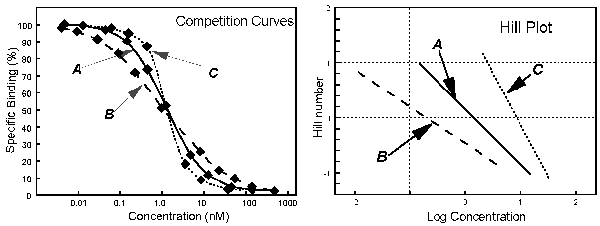
The slope factor describes the steepness of a curve. In most situations, there is no way to interpret the value in terms of chemistry or biology. If the slope factor is far from 1.0, then the binding does not follow the law of mass action with a single site. Often investigators transform the data to create a linear Hill plot. The slope of this plot equals the slope factor, and the intercept with an ordinate value of "0" equals the IC50.


Explanations for shallow binding curves include:
Often, data does not fit one site models well, and a two site analysis may be useful. (Normal experimental variance usually does not permit discrimination of two from three high affinity sites). Some assumptions needed to do such analyses include:
When you look at the competitive binding curve, you will only see a biphasic curve only in the rare cases where the affinities are very different. More often you will see a shallow curve with the two components blurred together. For example, this graph shows competition for two equally abundant sites with a ten fold (one log unit) difference in EC50. If you look carefully, you can see that the curve is shallow (it takes more than two log units to go from 90% to 10% competition), but you cannot see two distinct components.
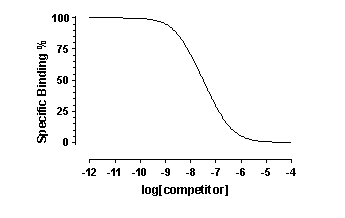
Some software (like Prism) can simultaneously fit your data to two equations and compare the two fits. This feature is commonly used to compare a one-site competitive binding model and a two-site competitive binding model. Since the model has an extra parameter and thus the curve has an extra inflection point, the two-site model almost always fits the data better than the one site model. And a three site model would fit even better. Before accepting the more complicated models, you need to ask whether the improvement in goodness of fit is more than you would expect by chance. Prism, for example, answers this question with an F test that yields a P value that answers this question: If the one site model were really correct, what is the chance that randomly chosen data points would fit to a two-site model this much better (or more so) than to a one-site model?
Before looking at Prism�s comparison of the two equations, you should look at both fits yourself. Sometimes the two-site fit gives results that are clearly nonsense. Here are some examples to when to disregard a two-site fit:
Basic Principle: If the results don�t make sense, don�t believe them (don�t ever say "The computer said ...."). Only pay attention to the results of the comparison when both fits are reasonable.
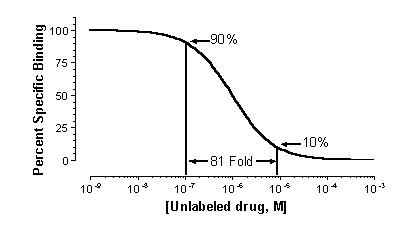
This equation has five variables: the top and bottom plateau binding, the fraction of the receptors of the first class, and the IC50 of competition of the unlabeled ligand for both classes of receptors. If you know the KD of the labeled ligand and its concentration, you can convert the IC50 values to Ki values.
If you use an antagonist radioligand and compete with an unlabeled agonist you are likely to observe a shallow competitive binding curve. Receptors can interact with intracellular molecules in a way that alters agonist binding. One situation that has been well studied is the competition of unlabeled agonist ligand for antagonist radioligands for binding to receptors linked to G proteins in membrane preparations lacking GTP.
The ternary complex model explains some of these data. This model shows binding of an agonist (A) to receptor (R) coupled to G protein (G):
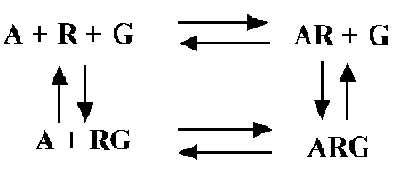
Some features of the model:
A few investigators have fit data to equations describing the ternary complex. Most do not, for these reasons:
Rather than fit data to the ternary complex model, most investigators fit data to a two-site binding model. The data fit well to the two site model, even though we know it is too simple. The two-site model is shown below - it is not the same as the ternary complex model.
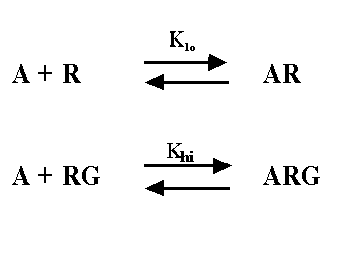
The two-site model, as its name implies, assumes that there are two distinct kinds of receptors. In this context, one kind of receptor is coupled to G, and the other is not. This simpler model provides values for Klo, Khi, and relative numbers of the two kinds of receptors, and these can be compared after different treatments. Since we know that the two-site model is too simple (the ternary complex is a better model for many systems) you shouldn't interpret Khi and Klo strictly. Nonetheless, the ratio of the Khi and Klo is a useful empirical measure which often correlates with agonist efficacy.
This short section is hardly adequate to explain the complexities of the ternary complex model, but should be sufficient to keep you from making common mistakes in interpretation.
The first widely used program in the field was "Ligand", a freely available, special-purpose nonlinear regression program for analyzing equilibrium radioligand binding data written by Munson, Rodbard, and collaborators (Methods in Enzymology 92:543, 1983). Today, there are many other alternatives to Ligand (for many different computer platforms) that may be easier to use or more facile with certain types of analyses.
