| Práctica
Eucariotas: gen humano |
| Vamos a analizar con diferentes herramientas de predicción de
genes una secuencia del genoma humano. Intentaremos identificar los exones
y los intrones, deducir la ORF del mRNA maduro, predecir si hay una región
promotora en la secuencia y decidiremos de entre todas las estructuras
génicas propuestas por las diferentes aplicaciones, cual nos parece
mejor. |
| Secuencia: |
>human
AGCTTTCTTCTTTTCCCTGTTGCTCAAATAAATAGTGTTCTTTGCTCAAA
CCCCCTTTCCCTCCTCCTTCTGCAATCTCAGCGCCTAGCGAAATCTGTTT
TCTTCATTGTAACCTCAGCTTCACCGCAATTAATTTTTTTTCCCTCTGGT
CACAAGATAATTCCTGACGCCAGTGAGTCTGGAGGTCAGACGAACAGCAA
ATTGGGGAACAAGGCGGCACTAATTCCTTACAAGTTCCTTGAAAAATCTT
TCGCTTAAAAAAAACGGGGGGTGGGGGGAGCTTCTTTGCTGTTCAGGGAT
TTATGCCTCGCGGAGCTGTGGCTCGAACCAGTGTTGGCTAAGGCGGACTG
GCAGGGGCAGGGAAGCTCAAAGATCTGGGGTGCTGCCAGGAAAAAGCAAA
TTCTGGAAGTTAATGGTTTTGAGTGATTTTTAAATCCTTGCTGGCGGAGA
GGCCCGCCTCTCCCCGGTATCAGCGCTTCCTCATTCTTTGAATCCGCGGC
TCCGCGGTCTTCGGCGTCAGACCAGCCGGAGGAAGCCTGTTTGCAATTTA
AGCGGGCTGTGAACGCCCAGGGCCGGCGGGGGCAGGGCCGAGGCGGGCCA
TTTTGAATAAAGAGGCGTGCCTTCCAGGCAGGCTCTATAAGTGACCGCCG
CGGCGAGCGTGCGCGCGTTGCAGGTCACTGTAGCGGACTTCTTTTGGTTT
TCTTTCTCTTTGGGGCACCTCTGGACTCACTCCCCAGCATGAAGGCGCTG
AGCCCGGTGCGCGGCTGCTACGAGGCGGTGTGCTGCCTGTCGGAACGCAG
TCTGGCCATCGCCCGGGGCCGAGGGAAGGGCCCGGCAGCTGAGGAGCCGC
TGAGCTTGCTGGACGACATGAACCACTGCTACTCCCGCCTGCGGGAACTG
GTACCCGGAGTCCCGAGAGGCACTCAGCTTAGCCAGGTGGAAATCCTACA
GCGCGTCATCGACTACATTCTCGACCTGCAGGTAGTCCTGGCCGAGCCAG
CCCCTGGACCCCCTGATGGCCCCCACCTTCCCATCCAGGTAAGCCTCGAA
GTCGGGACAGGGCTGAACACCCAGGCAAGGATGCTGCGGGACCCTCGGAG
CTCCCGATTGCCTCGCGTAACTCTTCCCTCTTTTCCTCTAATCAGACAGC
CGAGCTCGCTCCGGAACTTGTCATCTCCAACGACAAAAGGAGCTTTTGCC
ACTGACTCGGCCGTGTCCTGACACCTCCAGGTGAGTATCTCCTCTCTTGG
AGAGGGAGGTTTAAACGGCAAGTCCTGGAGTTGGCAGACGTTTTGAAAAA
TTGCCACTCACTCGGTTTAGGGAAACTGAGGCCAGAGAGGGACAAGTGAC
TTGCCCATGGTTGCATCAAATGAATGGCAGAGTCAGTTTCCATGTGATGT
GCATTTAAGCCTTAATGCGCCTGGCCCTGCCTCCGCAGTGGCCGAGGTCT
GGCAAGTAGACATGGTCCGACTAAATACAAGTCTTTCTGTTCCATGTTGT
ATAGGAGCTGTCTTCGGCAGCCCCCTCCCAGCTAGTGTCAATTCCAAGTA
GGAGGGGTAGCGCAACGTCCGCCTGTGGTCTTTGGCGCCAACTGGGTGGG
GGCAGCGTGGGGGGCGGAGTTATCAGGCTGGAGGTACAGACCAAGTTTCC
TCCCTGGCGCCGGCCAGTCTGCGGACGGCCCCCGCCTCGGCACGCTCGGC
GGAAACTGACTGCTCCTTGGTCTTCTTTCCTCCCCCGCCCAGAACGCAGG
TGCTGGCGCCCGTTCTGCCTGGGACCCCGGGAACCTCTCCTGCCGGAAGC
CGGACGGCAGGGATGGGCCCCAACTTCGCCCTGCCCACTTGACTTCACCA
AATCCCTTCCTGGAGACTAAACCTGGTGCTCAGGAGCGAAGGACTGTGAA
CTTGTGGCCTGAAGAGCCAGAGCTAGCTCTGGCCACCAGCTGGGCGACGT
CACCCTGCTCCCACCCCACCCCCAAGTTCTAAGGTCTTTTCAGAGCGTGG
AGGTGTGGAAGGAGTGGCTGCTCTCCAAACTATGCCAAGGCGGCGGCAGA
GCTGGTCTTCTGGTCTCCTTGGAGAAAGGTTCTGTTGCCCTGATTTATGA
ACTCTATAATAGAGTATATAGGTTTTGTACCTTTTTTACAGGAAGGTGAC
TTTCTGTAACAATGCGATGTATATTAAACTTTTTATAAAAGTTAACATTT
TGCATAATAAACGATTTTTAAACACTTGTGTATATGATGACACCCGTCTC
CATTAAGTACTAATGATGCTTTCTCGCACATGGCCGAATTTTGGGAGCTT
TGGGAAAGTGAACTTGCTTATTCTACGAGAGGGAAATGAAAAACTGCCTG
GTTGAGAGGGGATGGGGTGGAGAGAGAAGGGTTCATGATGGGAGTCTCAT
GTCCATTGAGGGATGGGTGCAGAGAAAAGTTCTGGCTCTGCCTCATTATT
TCAGAGATGAAACCAGAGACTGGTGCAAGCT
|
| Pasos: |
| Existen muchos servidores en los que correr aplicaciones para identificar
y modelar genes. Cada uno de ellos basa la búsqueda en propiedades
o algoritmos diferentes generando en ocasiones resultados distintos. En
general es conveniente consultar a varios de ellos para tener una idea
clara de la estructura génica de la secuencia. |
-
Veamos si hay evidencias de transcripción de algún fragmento
de nuestra secuencia. Para ello usaremos la herramienta BLAST contra
una base de datos de EST (Expresed Sequence Tag). En esta búsqueda
nos interesará el mejor alineamiento global posible de la secuencia
de EST con nuestra secuencia del gen. Lo ideal es encontrar un EST que
alinee en toda su longitud con nuestro gen con un e-value muy bajo.
- Entramos en la pagina web del BLAST del NCBI
y hacemos click en "Standard nucleotide-nucleotide BLAST [blastn]
- Copiamos nuestra secuencia y la pegamos en la caja de "Search"
- Elegimos la base de datos de "est" en el apartado "Choose database".
Como es una secuencia humana, elegimos la de ESTs de humanos
- El resto se deja como está y pulsamos "BLAST!"
Después de un rato debería salir un resultado semejante
a éste.
¿Qué significan, en el gráfico,
las líneas negras discontinuas que vemos entre las lineas rojas?
¿Existe alguna información ambigua
o contradictoria?
¿Qué podemos deducir de este resultado?
Sería conveniente ir haciendo un esquema en una hoja de papel,
en forma de mapa lineal, y representar los datos que vamos obteniendo.
¿Sería suficiente con este resultado
para conocer la estructura génica de nuestra secuencia?
Aunque podemos empezar a construir un modelo del gen a partir de
los resultados de BLAST contra ESTs: primero, existen ciertos datos contradictorios
y, segundo, no tenemos información sobre las fases abiertas de lectura.
Por tanto, vamos a usar una serie de servidores de predicción para
intentar confirmar el modelo que tenemos de momento, y para predecir codones
de inicio y terminación y regiones promotoras.
|
-
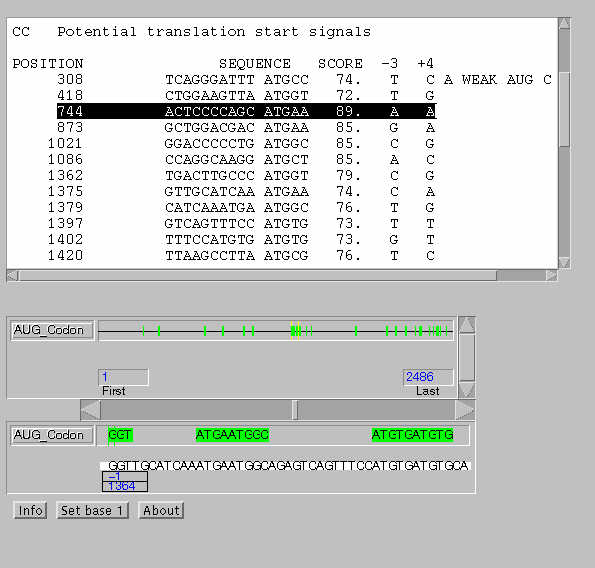
Intentemos identificar en la secuencia algún posible codon de
iniciación.
Abrimos, en un nueva ventana, el servidor AUG
evaluator.
A continuación elegimos la especie, insertamos la secuencia
en el campo correspondiente y presionamos "Submit" (resultado).
Identificar los codones de iniciación
predichos con una puntuación más alta.
¿Alguno concuerda con los datos que tenemos
hasta ahora?
|
-
Ahora vamos a usar varios servidores de predicción de exones
e intrones para identificar la fase abierta de lectura y validar nuestro
modelo. Esto es especialmente importante a la luz de los resultados
ambiguos obtenidos en el análisis con BLAST.
¿Hay diferencias entre los resultados
de los distintos programas?
¿Podemos validar el modelo propuesto a
partir de los resultados de BLAST?
|
-
Ahora vamos a repetir el proceso en un Metaserver. Los metaservers
son servidores que lanzan a su vez procesos en otros servidores de aplicaciones.
Algunos, simplemente, devuelven los resultados de todos los servidores
que han sido consultados y en otros integran las respuestas de forma más
o menos inteligente.
Nos conectamos a METAGENE,
abriendo una nueva ventana.
-
Escribimos nuestro correo electrónico en el campo correspondiente.
No es obligatorio pero si conveniente
-
Insertamos la secuencia en "Sequence"
-
Pasamos a "Sequence analysis options":
- Orientation y Features: todo seleccionado
- Engine Select; seleccionamos los servidores disponibles: GenScan,
Ebest y Grail.
- En Ebest: Human y Both seleccionados
- GeneScan: Vertebrate seleccionado
- Grail: Human seleccionado
-
Submit
Para ver los resultados hacemos click en el enlace "Current Search Results".
Esto nos llevará a una página donde tendremos la oportunidad
de ver el resultado de cada servidor en detalle. Lo más conveniente
es apretar donde pone "click here to annotate the results" Esto nos abre
una ventana donde compara los resultado de todos los servidores (resultado).
En el menu pinchamos en "Tools" y seleccionamos "Analysis" (resultado).
Aparece un histograma que permite valorar la
validez de la predicción consenso que genera el Metaserver.
Fijarse en los resultados de Ebest.
Nota: GeneBuilder
también es una herramienta muy completa de análisis. No consulta
a varios predictores de genes pero si busca ESTs y tiene un entorno gráfico
con mucha información. Es sencillo e intuitivo. Sería recomendable
probarlo con este ejemplo en algún momento libre.
|
-
Acabando: ¿Se puede detectar alguna isla GpC que coincida con
el principio del gen?
Para ello, analiza la secuencia genómica con CPGplot.
|
{kind=link}
{kind=link}
{kind=link}