Madrid-Jan-05
| 1. - Servers for
homology modelling |
2. - Other useful pages
|
3. - A walk through example
|
| 4. - Quick guide
for homology modelling |
SWISS-MODEL
SWISS-MODEL is an automated comparative modelling server (ExPASy, CH)
SDSC1
Protein structure homology modelling server (San Diego, USA)
3D-JIGSAW
Automated system for 3D models for proteins (Cancer Research UK)
WHATIF
WHAT IF Web interface: homology modelling, drug docking,
electrostatics calculations, structure validation and visualisation.
You have to provide your own alignments to build models.
ROBETTA
Robetta is a full-chain protein structure prediction server. It divides
protein chains into domains, and models them by either homology
modelling or ab initio modelling.
SQUARE
A server that will check how good your homology modelling or
fold recogniton alignment is (CNB, Madrid).
Loops Database
A table of five protein loop classes. (Cancer Research UK)
BIOTECH Validation Suite
An evaluation suite that uses three widely available validation
programs (PROCHECK, PROVE and WHAT IF - may not be available)
Verify3D
A tool designed to help in the refinement of crystallographic
structures. It also provides a visual analysis of model quality.
PDG-MAMMOTH
A local method for evaluating RMSD between model and the actual
target protein
A Homology Modelling Example -
a walk through exercise!!
|
:::: [BACK TO TOP] |
1.-This is the target sequence:
>target sequence
TQALVTLTSACADSSPSCDLFKNQGYCGEKFFDWMRKNCAKTCGFCGTGGGAETGGSDGVCGKTSVQQSR
IISGTNARPGAWPWMASLYMLSRSHICGGSLLNSRWILTASHCVVGTGATTKNLVIKLGEHDHYDKDGFE
QQFDVEKIIPHPAYKRGPLKNDIALIKLKTPARINKRVKTICLPKKGSAPSVGSRECYLAGWGSIRHPGG
SYHTLQQAMLPVVSYTNCHNQKNFVCAGFGKSSLTNACRGDSGGPLMCRKSDGSWEQHGIASFVVEYCKY
YTAFTPVANYIDWINQHINK
**** Note: Dont try this example because it almost certainly will not
work ...
!!! ****
2.- First: Submit the sequence to the SWISS-MODEL program:
We would need to use the "First approach mode", input your name and
e-mail address SWISS-MODEL
(First Approach mode). Check the advanced options - the type of
file output is very important for visualisation later.
After submission you would receive a confirmation message (and a
confirmatory e-mail= SEE an example
of it! ):
====================================
Then you'll receive an e-mail with all the proccesses conducted by
SWISS-MODEL
- SWISS-MODEL search for structural homologues. It uses "BLASTP2"
in
"ExNRL-3D" Database. Results are shown according to P(N) from BLAST
AlignMaster output
============================================================
Length of target sequence: 300 residues
Searching sequences of known 3D structures
Found
1bruP.pdb with P(N)=2e-40
Found
4chaB.pdb with P(N)=5e-38
Found
1choE.pdb with P(N)=5e-38
Found
1gmh_.pdb with P(N)=6e-38
Found
8gch_.pdb with P(N)=6e-38
Found
1gcd_.pdb with P(N)=6e-38
Found
1pytC.pdb with P(N)=6e-38
Found
3gch_.pdb with P(N)=8e-38
Found
5gch_.pdb with P(N)=8e-38
Found
4gch_.pdb with P(N)=8e-38
Found
1gmcA.pdb with P(N)=8e-38
Found
2gmt_.pdb with P(N)=8e-38
........ (cutfor clarity)
- With the "SIM" program all the "templates" (3D structures
used to model) with an id% > 25% more than 25 residues length will
be retrieved. In addition, this program can recognise different
domains.
For example ...:
"Extracting template sequences
"Running pair-wise alignments with target sequence
"Sequence identity of templates with target:
1bruP.pdb:44.98
% identity
4chaB.pdb:36.14
% identity
1choE.pdb:36.14
% identity
1gmh_.pdb:36.6
% identity
8gch_.pdb:36.6
% identity
1gcd_.pdb:36.6
% identity
1pytC.pdb:40.96
% identity
3gch_.pdb:30.78
% identity
5gch_.pdb:30.78
% identity
"Looking for template groups
"Global alignment overview:
Taget
Sequence:|==================================================|
1bruP.pdb|
--------------------------------------
4chaB.pdb|
---------------------------------------
1choE.pdb|
---------------------------------------
1gmh_.pdb|
---------------------------------------
8gch_.pdb|
---------------------------------------
1gcd_.pdb|
---------------------------------------
1pytC.pdb|
--------------------------------------
3gch_.pdb|
---------------------------------------
5gch_.pdb|
---------------------------------------
4gch_.pdb|
---------------------------------------
1gmcA.pdb|
---------------------------------------
2gmt_.pdb|
---------------------------------------
etc
etc...............................
AlignMaster found 1 regions to model separately: 1: Using template(s)
1a0lA.pdb
1a0lB.pdb 1a0lC.pdb 1a0lD.pdb
1a3bH.pdb 1a3eH.pdb 1a61H.pdb 1abiH.pdb
1acbE.pdb
1ad8H.pdb 1ae8H.pdb 1afeH.pdb
1ahtH.pdb 1ai8H.pdb 1aixH.pdb 1amhA.pdb
-------- ETC etc..
- Now batch files are created for ProMod to generate a model
Batch.1: residues 56 - 300 of submitted sequence.
Exiting AlignMaster
ProModII trace log for Batch.1
============================================================
ProModII: 3.70 (SP3)
ProModII: Loading Template: 1bruP.pdb
ProModII: Loading Template: 4chaB.pdb
ProModII: Loading Template: 1choE.pdb
ProModII: Loading Template: 1gmh_.pdb
ProModII: Loading Template: 8gch_.pdb
ProModII: Loading Raw Sequence
ProModII: Iterative Template Fitting
ProModII: Iterative Template Fitting
ProModII: Iterative Template Fitting
ProModII: Iterative Template Fitting
ProModII: Generating Structural Alignment
ProModII: Aligning Raw Sequence
ProModII: Refining Raw Sequence Alignment
ProModII: ProModII: doing complex assignment of backbone
ProModII: N-terminal overhang trimmed for chain ' '. Start at residue:
16
ProModII: ProModII: adding blocking groups
ProModII: Weighting Backbones
ProModII: Src residue is not amino-acid
ProModII: Src residue is not amino-acid
ProModII: Averaging Sidechains
ProModII: Adding Missing Sidechains
ProModII: Trying Ligating with anchor residues SER 32 and MET 35
ProModII: Trying Ligating with anchor residues SER 32 and LEU 36
ProModII: connectivity problem --> including residue LEU 22
ProModII: Trying Ligating with anchor residues SER 32 and SER 37
ProModII: connectivity problem --> including residue SER 23
ProModII: Trying Ligating with anchor residues SER 32 and ARG 38
ProModII: connectivity problem --> including residue ARG 24
ProModII: Trying Ligating with anchor residues SER 32 and SER 39
ProModII: Number of Ligations found: 500
ProModII: ACCEPTING loop 378: clash= 0 FF= 195.4 PP= -7.00
ProModII: Small Ligation (C-N < 3.0A) ignored;
ProModII: GROMOS will repair it at residue SER 25
ProModII: connectivity problem (C-N > 3.0A) at residue: 71
ProModII: Trying Ligating with anchor residues GLY 83 and GLN 86
ProModII: Number of Ligations found: 3
ProModII: all loops are bad; continuing CSP with larger segment
ProModII: Trying Ligating with anchor residues ASP 82 and GLN 86
ProModII: Number of Ligations found: 22
ProModII: all loops are bad; continuing CSP with larger segment
ProModII: Trying Ligating with anchor residues LYS 81 and GLN 86
ProModII: Number of Ligations found: 500
ProModII: ACCEPTING loop 230: clash= 0 FF= -17.5 PP= -2.00
ProModII: connectivity problem (C-N > 3.0A) at residue: 91
ProModII: Trying Ligating with anchor residues PRO 103 and ASN 106
ProModII: Number of Ligations found: 12
ProModII: all loops are bad; continuing CSP with larger segment
ProModII: Trying Ligating with anchor residues GLY 102 and ASN 106
ProModII: Number of Ligations found: 14
ProModII: all loops are bad; continuing CSP with larger segment
ProModII: Trying Ligating with anchor residues GLY 102 and ASP 107
ProModII: connectivity problem --> including residue ASP 93
ProModII: Trying Ligating with anchor residues GLY 102 and ILE 108
ProModII: Trying Ligating with anchor residues ARG 101 and ILE 108
ProModII: Number of Ligations found: 273
ProModII: ACCEPTING loop 218: clash= 0 FF= 1486.4 PP= 0.00
ProModII: Trying Ligating with anchor residues ASN 175 and ASN 178
ProModII: Trying Ligating with anchor residues HIS 174 and ASN 178
ProModII: Number of Ligations found: 22
ProModII: ACCEPTING loop 10: clash= 0 FF= -294.1 PP= -3.00
ProModII: Trying Ligating with anchor residues GLY 183 and LYS 186
ProModII: Trying Ligating with anchor residues ALA 182 and LYS 186
ProModII: Trying Ligating with anchor residues ALA 182 and SER 187
ProModII: Trying Ligating with anchor residues CYS 181 and SER 187
ProModII: Trying Ligating with anchor residues CYS 181 and SER 188
ProModII: Trying Ligating with anchor residues VAL 180 and SER 188
ProModII: Number of Ligations found: 500
ProModII: all loops are bad; continuing CSP with larger segment
ProModII: Trying Ligating with anchor residues VAL 180 and LEU 189
ProModII: Number of Ligations found: 112
ProModII: all loops are bad; continuing CSP with larger segment
ProModII: Trying Ligating with anchor residues PHE 179 and LEU 189
ProModII: Number of Ligations found: 500
ProModII: all loops are bad; continuing CSP with larger segment
ProModII: +++ Warning: Ligation Failed, SparePart will be inserted
later
ProModII: +++ It is usually the sign that the region is misaligned.
ProModII: connectivity problem (C-N > 3.0A) at residue: 177
ProModII: Trying Ligating with anchor residues LEU 189 and ALA 192
ProModII: Trying Ligating with anchor residues SER 188 and ALA 192
ProModII: Trying Ligating with anchor residues SER 187 and ALA 192
ProModII: Trying Ligating with anchor residues LYS 186 and ALA 192
ProModII: Trying Ligating with anchor residues GLY 185 and ALA 192
ProModII: Trying Ligating with anchor residues PHE 184 and ALA 192
ProModII: Trying Ligating with anchor residues GLY 183 and ALA 192
ProModII: Trying Ligating with anchor residues ALA 182 and ALA 192
ProModII: +++ Warning: Ligation Failed, SparePart will be inserted
later
ProModII: +++ It is usually the sign that the region is misaligned.
ProModII: connectivity problem (C-N > 3.0A) at residue: 207
ProModII: Trying Ligating with anchor residues VAL 219 and TYR 222
ProModII: Number of Ligations found: 7
ProModII: ACCEPTING loop 0: clash= 0 FF= -37.3 PP= 1.00
ProModII: Trying Ligating with anchor residues LYS 224 and THR 227
ProModII: Number of Ligations found: 2
ProModII: ACCEPTING loop 1: clash= 0 FF= 261.7 PP= 2.00
ProModII: Building CSP loop with anchor residues VAL 59 and ALA 64
ProModII: Number of Ligations found: 53
ProModII: all loops are bad; continuing CSP with larger segment
ProModII: Building CSP loop with anchor residues VAL 59 and THR 65
ProModII: Number of Ligations found: 119
ProModII: ACCEPTING loop 114: clash= 0 FF= 560.6 PP= 1.00
ProModII: Building CSP loop with anchor residues SER 136 and SER 139
ProModII: Building CSP loop with anchor residues PRO 135 and SER 139
ProModII: Building CSP loop with anchor residues ALA 134 and SER 139
ProModII: Number of Ligations found: 500
ProModII: ACCEPTING loop 22: clash= 0 FF= -38.9 PP= -2.00
ProModII: Finding Spare-Part loop with anchor residues ASN 178 and ASN
191
ProModII: connectivity problem --> including residue ASN 177
ProModII: Finding Spare-Part loop with anchor residues ASN 178 and ALA
192
ProModII: ACCEPTING loop 1 from 4RHV1 Clash= 4 FF= 555.6 PP=-29.69
ProModII: BadPhi= 1 BadGX= 0 BadXP= 0 weakXP= 0 Score= 7.00 rms= 0.00
ProModII: Optimizing Sidechains
ProModII: Dumping Preliminary Model
ProModII: Adding Hydrogens
ProModII: Optimizing loops and OXT (nb = 41)
ProModII: Final Total Energy: 5009.919 KJ/mol
ProModII: Removing Hydrogens
ProModII: Fixing Atom Nomenclature
ProModII: Dumping Sequence Alignment
***
Getting the 3D coordinates from the model
Finally you'll receive an
e-mail with the pdb coordinates if your model.
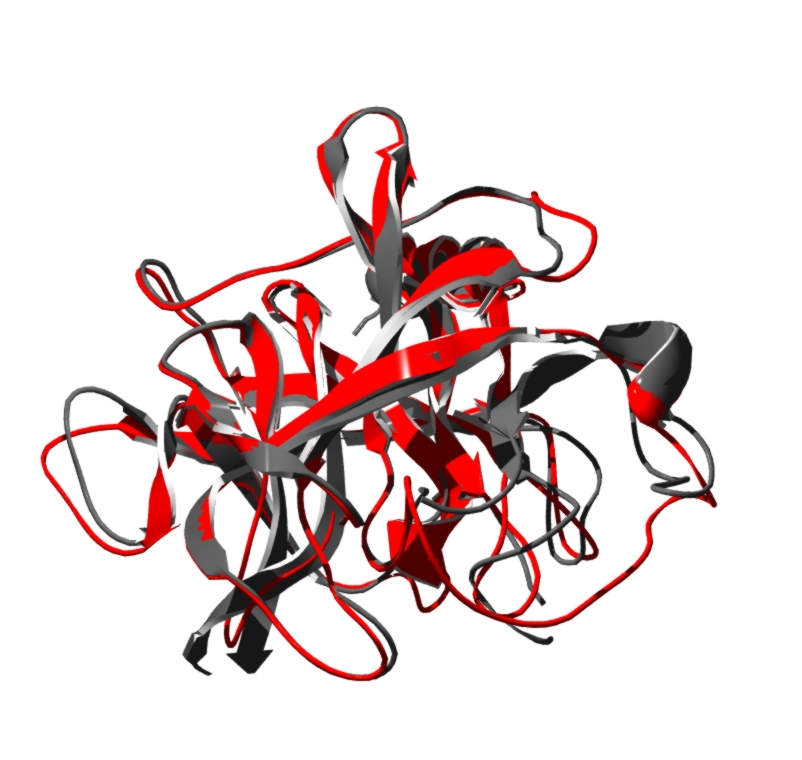
Here you can see in the image the model (RED) and one target (1bruP) in
grey.
These are predictions, so any conclusion must be taken carefully. (HERE
Access to an e-mail with the 3D
coordinates.).
Note that several templates have been selected to generate the model
although in the figure only one is represented. |
 |
An Example: Swiss
Pdb-Viewer to visualise the model
Example
2 Outputs: