![]()
In vitro pharmacology: concentration-response curves
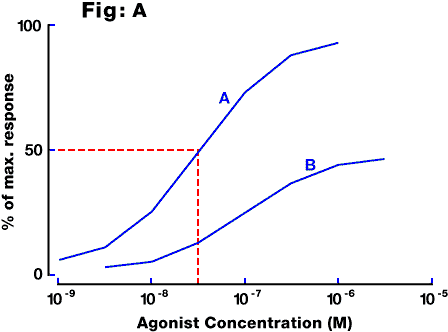
Fig. A: Examples of concentration-response curves in a typical in vitro preparation. A log concentration - response curve is usually sigmoidal, although the portion between 10% and 90% of maximal response approximates to a straight line and usually occurs over two orders of magnitude of concentration. Curve A is for a full agonist, with an EC50 of about 3x10(-8)M (intercept C). Curve B is for a partial agonist acting on the same receptor.
Agonist
A drug which binds to a receptor and activates it, producing a pharmacological response
(contraction, relaxation, secretion, enzyme activation, etc.).
Antagonist
A drug which attenuates the effect of an agonist. It may be competitive (or surmountable),
i.e. it binds reversibly to a region of the receptor in common with an agonist, but
occupies the site without activating the effector mechanism. The effects of a c ompetitive
antagonist may be overcome by increasing the concentration of agonist, thereby shifting
the equilibrium and increasing the proportion of receptors which the agonist occupies.
This type of antagonist is discussed further in the next section.
Alternatively, antagonists may be unsurmountable, where no amount of agonist can
completely overcome the inhibition once it has been established. Unsurmountable
antagonists may bind covalently to the agonist binding site (competi tive irreversible
antagonists), in which case there is a period before the covalent bond forms during which
competing ligands can prevent the inhibition. Other types of unsurmountable antagonists
act allosterically at a different site on the receptor or an associated ion channel. See
Jenkinson (1991) and Rang and Dale (1991) for more detail. Also see functional antagonism.
Competitive antagonist
See antagonist.
Desensitisation
See tachyphylaxis.
EC50
The molar concentration of an agonist which produces 50% of the maximum possible response
for that agonist. Other percentage values (EC25, EC40, etc) are
sometimes used.
Efficacy
A term introduced by Stephenson (1956) to describe the way in which agonists vary in the
response they produce even when they occupy the same number of receptors. High-efficacy
agonists can produce their maximal response while occupying a relatively low proportion of
receptors; agonists of lower efficacy cannot activate the receptors to the same degree and
may not be able to produce the same maximal response even when they occupy the entire
receptor population, thereby behaving as partial agonists. (See relative efficacy).
Functional antagonism
(or physiological antagonism). Reversal of the effects of a drug by an
agent which, rather than acting at the same receptor, causes a response in the tissue or
animal which opposes that induced by the drug. Examples include agents which have opposing
effects on an intracellular second messenger, or, in an
animal, on blood pressure. A functional antagonist can sometimes produce responses which
closely mimic those of the pharmacological kind.
Furchgott analysis
A method of measuring the affinity of an agonist by comparing its concentration-response
curve before and after inactivating a proportion of the receptors with an irreversible
antagonist. See Furchgott (1966) or Bowman and Rand (1980).
IC50
Where an agonist causes an inhibitory response, the IC50 is the molar
concentration which produces 50% of its maximum possible inhibition.
Insurmountable antagonist
Alternative name for unsurmountable antagonist (see antagonist).
Intrinsic activity
A term devised by Ariens in 1954 which attempted to describe the mathematical relationship
between receptor occupancy and tissue response. It has
now largely been replaced by efficacy, because the
definition of intrinsic activity means that it varies for a particular agonist between
different tissues, but efficacy, in theory, does not. However, intrinsic activity
is now widely used as an empirical measure of the maximal response to a test agonist as a
fraction of that to a full agonist of the same pharmacological class.
Inverse agonist
A drug which produces an effect opposite to that of an agonist, yet acts at the same
receptor. The best established examples act at the benzodiazepine receptor (see Schofield,
1989). Such compounds have also been described as negative antagonists,
or as having negative efficacy.
Irreversible antagonist
See antagonist.
KA
The dissociation equilibrium constant for an agonist. It is difficult to determine
experimentally, since it does not necessarily equal the concentration which produces
half-maximal response nor the concentration which occupies 50% of the receptors (see efficacy). It may be measured by Furchgott analysis;
alternatively, if assay conditions are identical, it may equal the Ki value determined in a binding assay. The
reciprocal is called the affinity constant or association constant. Do not confuse with
the physicochemical use of the same symbol. For more detailed information, see Jenkinson
(1989)
Occupancy
The proportion of receptors to which a drug is bound. It may be calculated from the
Hill-Langmuir adsorption isotherm:
[D]
Occupancy = ___________
K + [D]
where K is the dissociation constant for the drug and [D] is its concentration.
Partial agonist
An agonist which, no matter how high a concentration is applied, is unable to produce
maximal activation of the receptors. In a preparation with a low receptor reserve, it is therefore unable to produce a
maximal response. See also efficacy
pD2
The negative logarithm of the EC50 or IC50 value.
Potency
A measure of the concentrations of a drug at which it is effective. A much-abused, vague
term which should always be further defined. For agonists, EC50,
IC50, KA
or pD2 are usually used, while pA2, KB or pKB are used for antagonists. Other terms are used
in binding studies (see section 3) which do not distinguish
between agonists and antagonists. It is important to realise that the potency of an
agonist does not give any information about its affinity for the receptor, because the
pharmacological response is rarely directly proportional to receptor occupancy (see efficacy).
Receptor reserve
Because high-efficacy compounds need to occupy relatively few receptors to produce a
maximal response, it is possible to inactivate a proportion of the receptors in a tissue
(e.g. in the presence of an irreversible antagonist)
without depressing the maximum of the concentration-response curve. (The curve is,
however, shifted rightward along the x axis). There is said to be a receptor reserve (or,
more colloquially, spare receptors) for that particular agonist in that particular tissue.
There is no receptor reserve for a drug which acts as a partial agonist in the tissue. The
receptor reserve may vary between tissues, depending on the number of receptors in the
particular tissue and the efficiency of coupling between them and their effector
mechanism. Consequently, a partial agonist in one tissue may appear to act as a full
agonist in a tissue with a higher receptor reserve.
Relative efficacy
Stephenson (1956) originally proposed a numerical definition of agonist efficacy in which
a pure antagonist (i.e. one totally devoid of any agonist activity) was defined as having
zero efficacy, and a drug with an efficacy of 1 would, by definition, produce a maximal
response at full occupancy that was 50% of the maximal response to a high-efficacy
agonist. A more practical method of comparing agonist efficacy is to determine relative
efficacy, i.e. to compare the ratio of the receptor occupancy at which two agonists
produce the same response. There is no upper limit on the numerical value of efficacy or
relative efficacy. See intrinsic efficacy.
Relative potency
The ratio of the potency of a test drug (i.e. its EC50, IC50, etc.)
to that of a standard drug.
Second messenger
Intracellular substance (e.g. cyclic AMP or inositol phosphates), the concentration of
which may be controlled by activation of membrane receptors and which can control further
intracellular events (e.g. protein phosphorylation, neurotransmitter release or membrane
polarisation).
Selectivity
Relative potency of a drug between two receptor subtypes
for the same endogenous ligand. This is a relative rather than absolute term that should
always be qualified (e.g. prazosin is 30-fold selective for <28>1-adrenoceptors relative to <28>2-adrenoceptors).
Compare specificity.
Spare receptors
See receptor reserve.
Species homologue (or species variant)
A receptor for a particular neurotransmitter which mediates the same physiological
function in two species and is found in similar tissue locations. The two receptors differ
in amino-acid sequence to a small degree (approx. 10% or less), giving rise to differences
in the affinity of some antagonists or the relative potencies of agonists. Compare subtype.
Specificity
Relative potency of a drug between the receptors for two different endogenous ligands
(e.g. sulpiride is specific for dopamine receptors when compared
with 5HT receptors). Compare selectivity.
Subtype
Subtypes of receptor are those which, in a single species, are activated by the same
family of endogenous ligands but exhibit sufficient differences in their pharmacological
properties or molecular structure to justify being classified separately. Traditionally,
subtypes have been identified using drugs which can selectively activate them or
antagonise the effects of agonists with markedly different potencies (the usual rule of
thumb is that there should be at least a 10-fold difference in antagonist affinity, i.e.
one log unit difference in pKB value, when postulating the existence of a novel
receptor subtype (Kenakin et al., 1992)). Consequently, subtypes can only be
identified when pharmacological tools are available. Molecular biological techniques have
now determined the amino-acid sequence of many receptor proteins, and hence the degree of
homology between receptor types to be measured. However, there is no established rule
which differentiates receptor subtypes simply on the basis of the number of amino-acids
which differ between them. Compare species homologue.
Surmountable antagonist
See antagonist.
Tachyphylaxis
A reduction in the response to an agonist while it is continuously present at the
receptor, or a progressive reduction in the response upon repeated presentation of the
agonist.
Unsurmountable antagonist
See antagonist.
![]() Receptor
antagonism
Receptor
antagonism
![]() Physiological
salt solutions
Physiological
salt solutions
![]() Table of Contents
Table of Contents
 |
 |
 |
 |
 |
 |
 |