Análisis de secuencias:
más sobre búsquedas con perfiles. Práctica.
PSI-BLAST, PHI-BLAST y el
paquete HMMer.
1.-
PSI-BLAST
Para
ejecutar PSI-BLAST desde el servidor del NCBI hay que ir a http://www.ncbi.nlm.nih.gov/BLAST/
y
allí pinchar en "Protein Blast - PSI- and PHI-BLAST"
Si
lo tenéis instalado también podéis ejecutarlo localmente.
Como
es un poco lento (especialmente el servidor) en los siguientes ejemplos
los resultados están precalculados.
a)
YD33_MYCTU: Esta proteína está anotada como "Hypothetical
protein". Si hacemos un BLAST con ella obtenemos estos
resultados.
¿Nos
dicen algo BLAST acerca de su función?
La
secuencia de esta proteína es:
>YD33_MYCTU|Q10644|Hypothetical
protein Rv1333.
MNSITDVGGI RVGHYQRLDP DASLGAGWAC GVTVVLPPPG TVGAVDCRGG APGTRETDLL
DPANSVRFVD ALLLAGGSAY GLAAADGVMR WLEEHRRGVA MDSGVVPIVP GAVIFDLPVG
GWNCRPTADF GYSACAAAGV DVAVGTVGVG VGARAGALKG GVGTASATLQ SGVTVGVLAV
VNAAGNVVDP ATGLPWMADL VGEFALRAPP AEQIAALAQL SSPLGAFNTP FNTTIGVIAC
DAALSPAACR RIAIAAHDGL ARTIRPAHTP LDGDTVFALA TGAVAVPPEA GVPAALSPET
QLVTAVGAAA ADCLARAVLA GVLNAQPVAG IPTYRDMFPG AFGS
Podemos
hacer una búsqueda con PSI-BLAST
(o
con cualquiera de las herramientas que hemos visto anteriormente -Pfam,
Prosite, etc-) para intentar averiguar algo sobre esta proteína.
Como
el servidor de PSI-BLAST es demasiado lento para que lo usemos todos,
aquí
tenéis los resultados que obtendríais ejecutándolo
'localmente', por línea de comandos, con los parámetros
"-j 7 y -e 0.0001". Si queréis podéis ver la versión
'WEB' de la primera ronda,
de la segunda y de la tercera
ronda.
¿Proporciona información adicional
a BLAST? ¿Cuál creéis que podría ser la función
de la proteína (al menos podemos proponer una hipótesis sobre
la que trabajar)?
¿Qué
significa que las búsquedas convergieron?
Si quesieseis podríais buscar en Pfam.
Obtendríais esto.
b)
Búsqueda con una secuencia de la base de datos SCOP, correspondiente
al dominio: a.26.1.1: Granulocyte-colony stimulating factor
>d1bgc__:a.26.1.1
SLPQSFLLKCLEQVRKIQADGAELQERLCAAHKLCHPEELMLLRHSLGIPQAPLSSCSSQ
SLQLRGCLNQLHGGLFLYQGLLQALAGISPELAPTLDTLQLDVTDFATNIWLQMEDLGAA
PAVQPTQGAMPTFTSAFQRRAGGVLVASQLHRFLELAYRGLRYLA
El
resultado de hacer un PSI-BLAST localmente contra una base de datos no
redundante de swiss-prot, trembl y trembl_new generó estos resultados.
-Intentad comprender qué ha sucedido en las búsquedas
y qué nos ha permitido averiguar PSI-BLAST.
-En la ronda 2, aparece
la proteína "Q90YI0 (Q90YI0) Interleukin-6 precursor". Ésta
tiene una identidad del 20% con respecto a la "query", sin embargo tiene
un e-value bastante significativo (de 2e-06): ¿por qué?
-¿Qué
consecuencias tiene en la siguiente ronda la inclusión de esta secuencia
en el perfil?
-¿Por qué
en la última ronda las proteínas "interleukin" tienen mejores
e-values que las proteínas "granulocyte-..."?
Cuando queráis hacer un psi-blast local
tendréis que utilizar el programa blastpgp:
Funcionamiento más básico:
blastpgp -i fichero_input.fasta -o resultados.psiblast
-d base_de_datos_de_secuencias -e 1e-05 -j 5
·fichero_input.fasta: el fichero con la secuencia que queremos
analizar.
·resultados.psiblast: el fichero donde queremos
guardar los resultados de PSI-BLAST.
·base_de_datos_de_secuencias: una base de datos de
secuencias formateada con formatdb.
·"-e 1e-05": el umbral de e-value.
·"-j 5": el número de rondas o iteraciones,
cinco en este caso.
Para conocer las otras opciones del programa podéis poner "blastpgp
-". También tenéis información en el fichero README.bls
que viene con el programa.
2.-
HMMer
El
programa hmmpfam ya lo hemos probado a través de la web. Podríamos
probar a utilizar el programa hmmsearch con cualquiera de las secuencias
que hemos usado con PSI-BLAST. (El paquete de programas de
HMMer se puede descargar desde aquí).
Para ello (como ejemplo tomaremos el caso ya
visto de YD33_MYCTU):
-
Primero tendríamos que construir un HMM. Para
ello podríamos hacer un BLAST y hacer un alineamiento múltiple
con los homólogos resultantes (para esto es mejor usar el servidor
de BLAST del EMBL
ya que permite 'descargar' las secuencias de los homólogos en formato
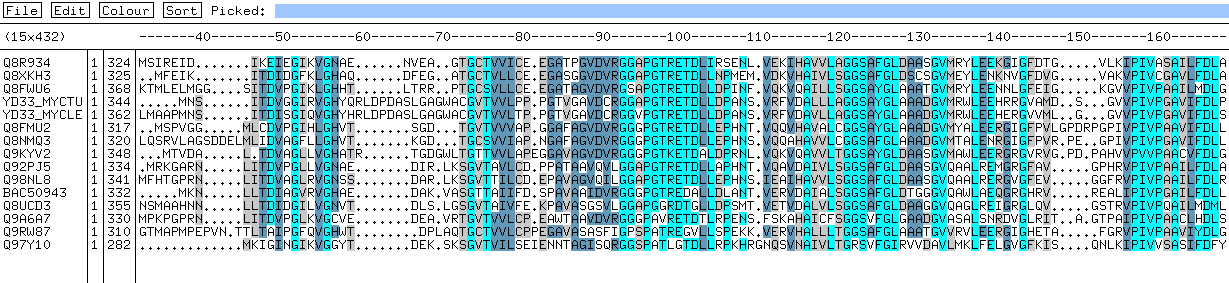
fasta). Alineamos estas secuencias con CLUSTAL. Con el alineamiento resultante
podemos construir un HMM con hmmbuild. Y finalmente, para calibrar
el HMM podemos ejecutar el hmmcalibrate.
-
Después podemos buscar en una base de datos
de secuencias usando el programa hmmsearch.
[resultado
de la búsqueda contra las secuencias swiss-prot+trembl+trembl_new
(930.860 secuencias); tardó más de 12 horas en una máquina
con dos procesadores]
**Para
obtener información de los parámetros de cada programa poner
"man hmmsearch" o "man hmmbuild"**
-
Con esos resultados podríamos volver a construir
un perfil-HMM e iniciar nuevas búsquedas. Lo primero que deberíamos
hacer es obtener las nuevas secuencias para hacer un nuevo alineamiento
múltiple. En este caso no tenemos una opción de "get selected
sequeces" así que podríamos hacerlo a mano o bien
hacer un programita en perl que extrajera esas secuencias, preferentemente
que extrajera la parte de las secuencias que se corresponde con el perfil-Hmm
y no las secuencias enteras.
{kind=link}