
|
 |
HAMAP documentation
HIGH-QUALITY
AUTOMATED AND
MANUAL
ANNOTATION OF MICROBIAL
PROTEOMES
|
|
 |
Et microbes plus merveilleux
que les sept merveilles du monde
(G. Apollinaire)
See also a presentation of HAMAP in slides
and the
HAMAP links page.
Citation
A. Gattiker, K. Michoud, C. Rivoire, A. H. Auchincloss, E. Coudert, T. Lima, P. Kersey,
M. Pagni, C.J.A. Sigrist, C. Lachaize, A.-L. Veuthey, E. Gasteiger, A. Bairoch.
"Automatic annotation of microbial proteomes in Swiss-Prot". Comput. Biol. Chem. 27:49-58(2003).
doi:10.1016/S1476-9271(02)00094-4
If you have any questions or suggestions regarding the HAMAP project and
data, please send us email at: hamap@isb-sib.ch.
[General]
[Accessing HAMAP]
[Content of a HAMAP family]
[Availability of HAMAP]
[Technical aspects]
[Links]
[Frequently asked questions]
 General
General
What is HAMAP?
HAMAP stands for High-quality Automated and Manual Annotation of microbial Proteomes.
The HAMAP project aims to automatically annotate in SWISS-PROT a significant percentage of proteins
originating from bacterial and archaeal genome sequencing projects, with no decrease in
quality. It is also used to annotate proteins encoded by complete plant and algal plastid
genomes (chloroplasts, cyanelles), and will be extended to mitochondrial genomes.
Our automatic annotation methods, using a rule-based system, are only applied in the
cases where they are able to produce the same quality as manual annotation would. This
concerns two distinct subsets of proteins:
- proteins that have no significant similarity to any other microbial or non-microbial proteins (ORFans);
- proteins that are part of well-defined families or subfamilies.
Many checks are enforced in order to prevent the
propagation of wrong annotation and to spot problematic cases, which are
channeled to manual curation. The results of this annotation are integrated
in SWISS-PROT, and a website is provided at
http://www.expasy.org/sprot/hamap/.
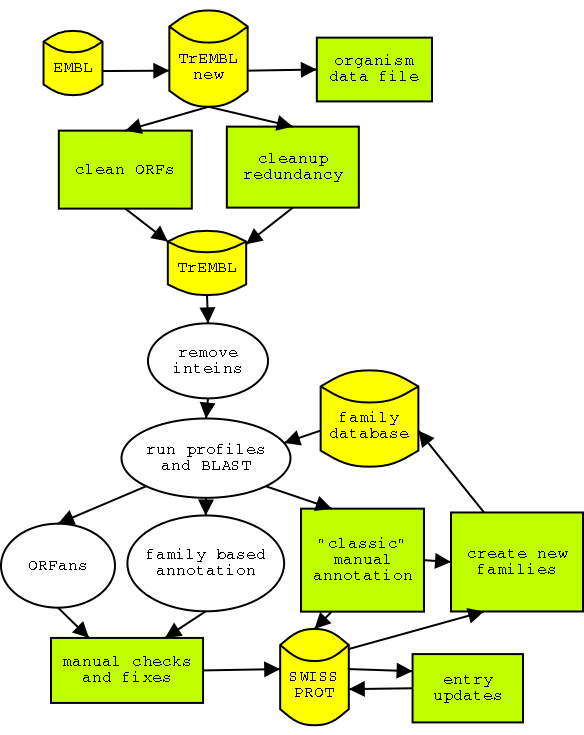
The structure of the HAMAP annotation pipeline. Manual
and partly automated steps are in green. The automated annotation part is in white.
Why HAMAP?
Between 1995 and 2002, over 100 microbial genomes have been entirely sequenced;
they encode over 200,000 protein sequences. The classical manual annotation
methods used in SWISS-PROT are not applicable here, despite the considerable
demand for corrected and annotated complete proteome sequences. So, we have
developed a semi-automatic, rule-based method for the high-quality annotation
of subsets of microbial proteomes.
See the HAMAP status report
page for up-to-date statistics about the number of complete microbial proteomes
currently available.
HAMAP participants
This project principally involves the SWISS-PROT group at SIB in Geneva. It is
also developed in collaboration with other groups of the SIB, the SWISS-PROT
group at EBI, and with the groups of François Rechenmann and Alain Viari
(Grenoble) and Laurent Duret and Guy Perrière (Lyon).
Copyright notice
HAMAP is copyright by the Swiss Institute of Bioinformatics. It is a part of
the SWISS-PROT protein knowledgebase. See http://www.isb-sib.ch/announce/ for
the license terms.
Content of a HAMAP family
An example of a HAMAP family can be found at http://www.expasy.org/cgi-bin/hamap/nicemf.cgi?MF_00163.
The view of each family rule contains:
-
Annotation that is propagated to member entries (e.g. protein name,
comments, keywords) with the extent of the propagation.
-
Computed features (e.g. export signals, transmembrane regions) that may
be applied to entries by using appropriate prediction programs.
-
Characteristics of the family (e.g. fusion, duplication, size range).
-
Comments on the family.
-
Alignments of a representative set of entries. There is usually a single
alignment per family, unless there is a 'deep split' between bacterial
and archaeal entries.
-
Sets of member sequences.
-
Taxonomic distribution of member sequences in SWISS-PROT and TrEMBL hits
from complete proteomes.
-
The possibility to display the NiceProt View of SWISS-PROT of any
microbial protein from any species belonging to the family.
Accessing HAMAP
The most efficient
and user-friendly way to access HAMAP data
is to browse interactively on one
of the
mirror sites
of the ExPASy server, at http://www.expasy.org/sprot/hamap/.
Content of HAMAP data
The HAMAP release is concurrent with every SWISS-PROT weekly update. New
families are added in each release, and existing
families are periodically updated.
See the HAMAP families
page for up-to-date statistics about the number and coverage of HAMAP families.
From this page, it is also possible to browse and perform searches in HAMAP families,
and to scan a user-entered sequence.
Cross-references from SWISS-PROT to HAMAP
Cross-references are present in all SWISS-PROT entries that are members of a
HAMAP family (or several). These cross-references take the form of a DR line
in the following format:
DR HAMAP; family-identifier; status; count.
The identifiers of the DR line are:
| family-identifier |
HAMAP unique identifier for a microbe protein family |
| status |
The values are either '-', 'fused', 'atypical' or 'atypical/fused'. The value '-' is a placeholder for an
empty field; the 'fused' value indicates that the family rule does not cover the entire protein; the value
'atypical' points out that the protein is divergent in sequence or has mutated functional sites, and should not be included in family datasets. The value 'atypical/fused' indicates both latter findings. |
| count |
Number of domains found in the protein, generally '1', rarely '2' for the fusion of 2 identical domains. |
| Example: |
DR HAMAP; MF_00012; -; 1. |
Availability of HAMAP
Downloading HAMAP data
- Complete proteomes. For each microbial proteome, a
complete proteome set is compiled and contains entries from SWISS-PROT and
TrEMBL. These sets are non-redundant and contain all known proteins encoded in
each proteome, including sequence corrections and annotation of newly
discovered genes. They are available in SWISS-PROT and FASTA format at
http://www.expasy.org/ftp/databases/complete_proteomes/. Alternatively, the
sets can be retrieved by using the keyword Complete proteome in the Sequence
Retrieval System (SRS, http://www.expasy.org/srs/).
- Proteome analysis. Statistical and comparative analyses of complete proteomes,
compiled from SWISS-PROT, TrEMBL, InterPro, CluSTr and GO, is available from
the EBI Proteome Analysis pages.
- HAMAP families. Families are currently available online on ExPASy but they are not distributed, as they are used for
internal purposes in the SWISS-PROT database and their format is subject to
frequent and unannounced changes. However we are looking forward to
collaborating with interested users - please send enquiries to hamap@isb-sib.ch.
Linking to HAMAP
See How to create HTML
links to services on ExPASy to find out how to create links to HAMAP web pages.
Technical aspects
How to identify the result of automatic annotation
Every SWISS-PROT entry incorporates annotation extracted from a variety of
information sources, and it is not currently possible to mark the origin(s) of
each annotation item in the database. The objective of the database lies more
in providing a homogeneous view of the data.
Automatically annotated entries present these general features:
The extent of the annotation that is propagated automatically can be found
in each family rule.
How HAMAP data is maintained
A relational database has been developed to store family rules, profiles,
sequences and hits which supports incremental updates. The HAMAP profiles
are generated using an automatic procedure based on the method used
to generate PROSITE profiles (see Sigrist et al.,
Brief. Bioinform. 3(3):265-274 (2002)).
ORFan annotation procedure
Proteins with no identified domains (in PROSITE, Pfam, etc.) and with no
significant BLAST similarities except in very close species undergo basic
annotation as hypothetical proteins, with possible features predicted by
sequence analysis programs. The ORFans module is run on such entries to:
-
Create an ID, DE and GN line (see the SWISS-PROT user manual);
-
Run programs to detect inteins (protein splicing), signal sequence,
transmembrane regions, coiled coils, some repeats (ANK, WD), ATP/GTP
binding sites, LPXTG motifs (cell-wall anchor);
-
Based on the above results, create the relevant keyword, feature
and comment lines;
-
Take into account rules of exclusion, dependency, consistency (e.g.
transmembrane regions cannot be present if the protein is supposed to
be secreted).
Feature propagation
Protein features (and associated comments and keywords) are propagated automatically
using two different approaches.
General features are propagated on the basis of their
conservation throughout the family. The alignment(s) of representative entries
present in the family rule is used to transfer features from the family rule to
new members, provided that conserved residues specified in the family are
observed.
Computed features are predicted using ad hoc methods:
Links
Links to microbe-specific resources
See the
HAMAP links page
SWISS-PROT documents relevant to Bacteria and Archaea
Frequently asked questions
How will the annotation coherence be checked?
This module is being developed. It should be able to warn annotators about
missing proteins, problems of orthologues and paralogues, proteins belonging to
a pathway not supposed to exist in a given organism. It will use the
information provided by rules specific to the organisms
themselves and to the biological pathways.
Will HAMAP be extended to eukaryotes?
HAMAP is already used to annotate the simple genomes of plastids and, in the
future, of mitochondria. The HAMAP annotation procedure relies on a very
high quality of gene prediction in genome sequences. While this postulate is
true for most submitted prokaryotic genomes, the complex structure of
eukaryotic genes makes high-quality automatic annotation very difficult.
However, we are extending the concepts developed in HAMAP in a new
system under development to assist curators' work based on PROSITE
entries.
What is the coverage of HAMAP in a genome?
Since family rules have been
built with a bias toward well-studied phyla and housekeeping genes, the
coverage is dependent on the organism type and the genome size. HAMAP families cover
40% of the genome in Buchnera aphidicola (subsp. Acyrthosiphon
pisum), 11% in Escherichia coli K12, and only 4%
in Streptomyces coelicolor.
The number of ORFans per genome is also very variable, from virtually none in Buchnera aphidicola to 40% in Mycobacterium tuberculosis.
Is it possible to annotate all the proteins of a new complete genome by HAMAP?
In certain new genomes, it is possible to annotate over a third of the
proteins automatically with the current set of families and the ORFans module.
This coverage is constantly expanded with the addition of new families.
However, the current approach is intrinsically limited to 'well-behaved' orthologous
families, and new methods are being developed for the annotation of complex
protein families.
Who are those colored beasts appearing at the top of each page?
Archie, Becky and Chloe are the HAMAP mascots.
They are guaranteed to be non-pathogenic, hypoallergenic, and organically cultured.
 ExPASy Home page
ExPASy Home page